Хроматография
Хроматографией называют метод разделения веществ, основанный на различном взаимодействии компонентов смеси с подвижной и неподвижной фазами в динамических условиях. В процессе хроматографирования подвижная фаза (жидкость или газ), содержащая анализируемую пробу, перемещается через неподвижную. При этом происходит многократное повторение актов взаимодействия, что обусловливает высокую эффективность разделения веществ с близкими свойствами. Во всех случаях компоненты разделяемой смеси распределяются между подвижной и неподвижной фазами, и их перемещение по колонке или по листу бумаги протекает с различной скоростью. В результате достигается разделение.
Подвижную фазу, проходящую через неподвижную, часто вызывают элюентом; растворенные вещества, покидающие неподвижную фазу вместе с элюентом, - элюатом и, наконец, процесс перемещения разделяемых компонентов образца вместе с элюентом - элюированием.
В колоночной хроматографии обычно проводят элюирование до тех пор, пока разделенные компоненты смеси не выйдут один за другим из колонки. Фракции элюата, в которых содержатся разделенные компоненты, собирают отдельно и анализируют соответствующими аналитическими методами.
В современных хроматографах - приборах для проведения хроматографического разделения и анализа - для определения состава и содержания разделенных компонентов смеси после выхода из колонки установлены детекторы.
Детекторы регистрируют изменение физических свойств элюата; их показания можно с помощью самописца записывать автоматически, получая при этом кривую элюирования - хроматограмму. Каждый пик на такой кривой (рис. 175) соответствует отдельному компоненту, а площадь каждого пика характеризует относительное содержание данного компонента. Основными характеристиками пиков являются удаленность их центров тяжести от точки, отмечающей момент ввода образца в хроматограф, а также ширина.
Положение пиков можно характеризовать либо временем выхода соответствующих компонентов образца, либо удерживаемым объемом VR.
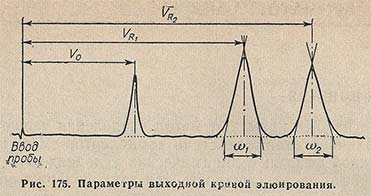
Степень хроматографического разделения обычно оценивают отношением расстояния между максимумами пиков двух веществ к сумме их полуширин. Для оценки в равной мере важны как расхождение пиков, так и их уширение.
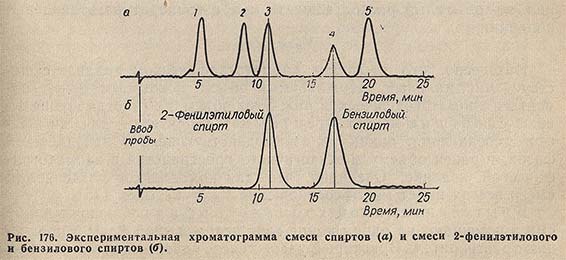
Идентификацию осуществляют хроматографированием стандартных веществ (реперов) и отождествлением их удерживаемых объемов с удерживаемыми объемами компонентов исследуемой смеси. Так, при сопоставлении хроматограммы анализируемой смеси спиртов (рис. 176) с хроматограммой смеси 2-фенилэтилового и бензилового спиртов видно, что в анализируемой смеси содержатся оба эти спирта.
Для количественного определения разделенных компонентов зависимость величины сигнала детектора (т. е. отклонения показаний самописца или прибора индикатора) от концентрации компонента устанавливают калибровкой. При калибровке фиксируют изменение высоты пика или его площади от количества стандартного вещества, подаваемого в колонку. Высоту пика от вершины до нулевой линии измеряют линейкой. Площади пиков определяют несколькими способами: планиметром, электронным интегратором, взвешиванием бумаги, вырезанной из хроматограммы по контурам кривой, образующей пик, и умножением высоты пика на его ширину (на расстоянии, равном половине высоты от основания). Ширину узких пиков можно измерить с помощью специальной измерительной лупы.
Хроматографические методы используются как в аналитических, так и в препаративных целях. В первом случае с их помощью определяют состав смесей, контролируют чистоту препаратов, идентифицируют вещества, очищают их от примесей и т. п. В качестве препаративного метода разделения хроматографию используют для выделения и очистки природных веществ или продуктов химических реакций.
Существует много вариантов хроматографического анализа. Хроматографические методы различают по агрегатному состоянию фаз, между которыми происходит разделение компонентов смеси, по методике эксперимента и механизмам разделения.
В газовой хроматографии применяют системы газ - жидкость и газ - твердое вещество. Жидкостная хроматография включает системы жидкость - жидкость и жидкость - твердое вещество.
По механизму разделения хроматографические методы обычно подразделяются по типу взаимодействия компонентов смеси с сорбентом: адсорбционная хроматография - на поверхности сорбента, распределительная - в объеме сорбента.
Метод разделения, в котором в качестве твердой фазы используются ионообменные материалы, называют ионообменной хроматографией.
Осадочная хроматография основана на химических реакциях хемосорбента с компонентами смеси растворенных веществ с образованием новой фазы - осадка.
В эксклюзионной (гель-проникающей, ситовой) хроматографии используется способность материалов с контролируемой пористостью «сортировать» и разделять компоненты смеси в соответствии с размером и формой их молекул.
Разделение компонентов смеси может осуществляться в колонках (колоночная хроматография) и без них. К неколоночным методам относятся бумажная и тонкослойная хроматография.
Процесс переноса растворенных веществ подвижной фазой через неподвижную называют проявлением. (Не следует путать проявление с опрыскиванием тонкослойных хроматограмм реагентами, которые образуют с бесцветными компонентами окрашенные производные и таким образом раскрывают присутствие разделенных компонентов. Этот процесс часто также называют проявлением, хотя его лучше называть обнаружением, выявлением или детектированием.)
Проявление можно вести тремя различными методами: элюированием, фронтальным анализом, вытеснением. Наиболее широко используется элюирование.
При фронтальном анализе раствор исследуемой смеси веществ непрерывно подают к одному концу неподвижной фазы и заставляют ее продвигаться к другому концу. Если исследуемый раствор состоит из смеси компонентов А и Б в несорбируемом растворителе В, то первым из слоя начинает вытекать чистый растворитель В. После насыщения слоя сорбента менее сорбирующимся компонентом А из колонки будет вытекать раствор А в растворителе В. Наконец, когда сорбент насытится веществом Б, из колонки будет вытекать раствор компонентов А и Б, т. е. смесь исходного состава. Таким образом, фронтальным методом можно получить в чистом виде только менее сорбируемый компонент А.
Фронтальный метод применяется для очистки от примесей, если эти примеси сорбируются значительно лучше, чем очищаемое вещество. Для аналитических целей он неприменим.
При элюировании (или элюентном методе хроматографирования) исследуемую смесь (например, компонентов А и Б) растворяют в подходящем растворителе и вводят в верхнюю часть колонки или в начальный участок бумажной полоски.
При этом компоненты А и Б практически не разделяются или разделяются в очень малой степени. Если через неподвижную фазу пропускать подвижную (растворитель или смесь растворителей), которая слабее удерживается твердой фазой, чем компоненты А и Б, то последние будут вымываться, т. е. перемещаться вниз по колонке (слою, бумаге). Скорость этого перемещения зависит от концентрационных коэффициентов распределения компонентов, называемых в современной хроматографии факторами емкости К:

Чем меньше К растворенного компонента смеси, тем быстрее он движется вниз по колонке (слою, бумаге). Даже при невысоком различии коэффициентов распределения компоненты А и Б постепенно разделяются, а их зоны перекрываются все меньше по мере перемещения по колонке. Чистые вещества А и Б собирают по выходе из колонки (слоя, бумаги).
В колоночной хроматографии объем элюента, необходимый для извлечения из колонки максимальной концентрации вещества, называют объемом удерживания VR.
Время от момента ввода пробы в колонку до момента выхода из нее максимальной концентрации определяемого вещества называют временем удерживания tR.
Объем удерживания VR равен произведению времени удерживания на объемный расход элюента w при температуре и давлении колонки:
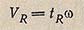
Интересно, что в любом хроматографическом анализе существует минимальный удерживаемый объем, с которым может появиться хроматографический пик. Этот объем V0 (иногда его называют свободным, или «мертвым») характерен для химического соединения, никак не взаимодействующего с неподвижной фазой, и равен объему межчастичного пространства в хроматографической колонке. Очевидно, что для компонентов разделяемой смеси всегда имеет место соотношение VR > V0 (рис. 175).
В вытеснительной хроматографии небольшую пробу смеси помещают на одном конце слоя неподвижной фазы и пропускают растворитель В, который удерживается твердой фазой сильнее, чем компоненты А и Б. Растворитель В вытесняет сорбированные вещества А и Б и проталкивает их по всему слою неподвижной фазы. Если компонент Б удерживается неподвижной фазой сильнее, чем компонент А, то Б вытесняет А и последний движется вниз по колонке первым; при этом образуются зоны от чистого А до чистого Б и от чистого Б до чистого В. Вытеснительный метод обычно используют для разделения больших количеств веществ и получения чистых соединений.
Газовая хроматография
В этом методе разделения подвижной фазой служит газ (гелий, азот), протекающий через неподвижную фазу.
Различают две принципиальные разновидности газовой хроматографии: в системе газ - твердое вещество (адсорбционная хроматография) и в системе газ - жидкость (газо-жидкостная хроматография). В первом случае разделение происходит за счет адсорбции веществ на поверхности твердого адсорбента, которым наполнена хроматографическая колонка. Во втором случае анализируемая газовая смесь проходит через колонку, наполненную твердым носителем (определенной степени зернения), на поверхность которого нанесен тонкий слой нелетучей жидкости. Эффективность разделения в газо-жидкостной хроматографии определяется не процессами сорбции - десорбции газа, а степенью растворения газообразных компонентов анализируемого вещества в жидкой нелетучей пленке. В качестве жидкой фазы в газо-жидкостной хроматографии используют вазелиновое масло, силиконовое масло, эфиры фталевой кислоты, трикрезилфосфат и др. В качестве твердых носителей применяют инертные вещества с развитой поверхностью, но малой микропористостью, чтобы исключить адсорбцию газа на поверхности. Наибольшее распространение получили каолин, диатомиты, тефлон и др.
При хроматографии в системе газ - адсорбент используют приемы фронтальной, элюентной и вытеснительной хроматографии; в системе газ - жидкость применяют только метод элюирования.
В настоящее время методами газовой хроматографии можно выполнять качественные и количественные определения компонентов смесей органических и неорганических газообразных, жидких и твердых веществ, давление паров которых превышает 0,133-133 Па, перегоняющихся без разложения в области температур до 400-500 °С. Основными достоинствами газовой хроматографии являются высокая чувствительность и разделяющая способность, скорость, точность и высокая степень автоматизации анализа.
Аппаратура для газовой хроматографии
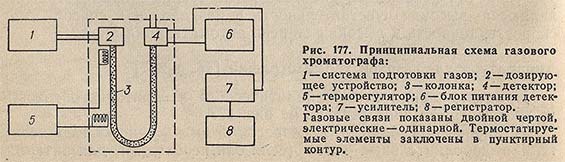
Принципиальная схема аналитического лабораторного газового хроматографа представлена на рис. 177.
Установка и стабилизация скорости потока и очистка газа-носителя и дополнительных газов (если они необходимы для питания детектора), а также измерение скорости потока газа выполняются системой подготовки газов 1.
Особенно важное значение имеет установка и стабилизация расхода газа-носителя, оказывающего непосредственное влияние на параметры удерживания и размеры пиков. Для этих целей используют совокупность нескольких элементов, основными из которых являются дроссель, регулятор давления и регулятор расхода. С помощью дросселя регулируют расход (объемную скорость) газа, изменяя аэродинамическое сопротивление канала, по которому этот газ течет. Если питание газового хроматографа осуществляется от индивидуального баллона газом, то дроссель обеспечивает и необходимый постоянный расход газа. В общем же случае используют регулятор давления, стабилизирующий давление на входе в хроматографическую колонку.
В некоторых случаях, например при программировании температуры колонки, необходимо поддерживать постоянный расход газа-носителя через колонку, когда ее сопротивление изменяется в процессе анализа. Для этой цели используют регулятор расхода. Он реагирует на изменение сопротивления колонки изменением сопротивления входящего в его состав регулирующего дросселя, так что суммарное сопротивление остается неизменным и расход не меняется.
Величины газовых потоков измеряют с помощью мыльно-пленочных измерителей по времени прохождения мыльной пленкой известного объема калиброванной стеклянной трубки.
Дозирующее устройство 2 позволяет вводить в поток газа-носителя непосредственно перед колонкой определенное количество анализируемой смеси в газообразном состоянии. Поток газа-носителя вносит анализируемую пробу в колонку 3, где осуществляется разделение смеси. Составляющие ее компоненты вместе с газом-носителем подаются в детектор 4, который преобразует разницу в физических или физико-химических свойствах бинарных смесей компонент - газ-носитель по сравнению с чистым газом-носителем в электрический сигнал. Величина сигнала зависит как от природы компонента, так и от содержания его в анализируемой смеси.
Необходимые температурные режимы колонки, детектора и дозирующих устройств достигаются помещением их в термостат, управляемый терморегулятором 5.
Сигнал детектора, преобразованный усилителем 7, записывается в виде хроматограммы автоматическим потенциометром 8.
Количественная обработка хроматограмм может производиться вручную или с помощью интегратора, автоматически фиксирующего площадь пика и время удерживания.
Существует несколько типов дозаторов. Для дозирования газообразных смесей используют газовые краны-дозаторы, позволяющие включать калиброванную емкость, предварительно заполненную анализируемой газовой смесью, в поток газа-носителя, который переносит дозу в виде газовой «пробки» в хроматографическую колонку. Жидкие смеси вводят в колонку специальными шприцами объемом 0,1-50 мкл через термостойкое резиновое уплотнение испарителя. (Испаритель представляет собой нагреваемый до определенной температуры металлический блок с каналом для ввода и испарения жидкой пробы. В канал подается поток предварительно нагретого газа-носителя. С одной стороны канал закрыт пробкой из термостойкой резины, с другой присоединена хроматографическая колонка.) Твердые вещества чаще всего вводят в виде раствора. При этом растворитель должен не только хорошо растворять твердые компоненты пробы, но и образовывать пик, не мешающий измерению пиков анализируемых компонентов.
Хроматографические колонки
Различают три основных типа аналитических колонок - насадочные (набивные), микронасадочные и капиллярные. Ввиду простоты приготовления наибольшее распространение получили насадочные колонки.
У насадочной колонки внутренняя полость заполнена инертным твердым носителем, покрытым тонкой пленкой нелетучего растворителя (неподвижной фазы) или твердым активным веществом (адсорбентом). Эти колонки изготавливают из металлических (нержавеющая сталь, медь), стеклянных или фторопластовых трубок с внутренним диаметром 2-10 мм (чаще 2-4 мм) и длиной 0,5-3,0 м. Им придают спиральную, U-образную или зигзагообразную форму. Микронасадочные колонки отличаются от насадочных только диаметром трубок (0,8-1,0 мм). Форма колонок практически не влияет на эффективность разделения и определяется размерами термостата.
В капиллярных колонках неподвижная фаза не заполняет всю внутреннюю полость, а покрывает лишь внутреннюю поверхность трубки, остающейся по существу полой и после нанесения неподвижной фазы.
Заполнение прямых и U-образных колонок сыпучим материалом не представляет трудностей. Наполнитель насыпают в колонку (через воронку) и осторожным постукиванием добиваются равномерного распределения его по длине колонки. Важно, чтобы столбик наполнителя не имел разрывов и чтобы отсутствовали местные пустоты. В спиралеобразные колонки рекомендуется насыпать наполнитель порциями с одного конца. Медленно поворачивая колонку по окружности спирали при одновременной ее вибрации с помощью касания с вращающимся эксцентриком, насаженным на ось моторчика, переводят наполнитель в другой конец колонки. Операцию повторяют до полного заполнения. Оптимальная плотность наполнения составляет примерно 0,3 г/см3.
При заполнении капиллярных колонок раствор жидкой фазы в летучем растворителе (эфире, пентане) продавливают из сосуда с двумя отверстиями с помощью сжатого газа (азота) через колонку до появления жидкости на противоположном конце. Затем сосуд отсоединяют и продувают колонку газом до полного удаления паров растворителя.
Детекторы
В газовой хроматографии наибольшее распространение получили ионизационно-пламенный детектор и катарометр (детектор по теплопроводности).
Катарометр измеряет различие в теплопроводности чистого газа-носителя и его смеси с веществом, выходящим из колонки. Поэтому чувствительность тем выше, чем больше теплопроводность анализируемого вещества отличается от теплопроводности газа-носителя. Подавляющая часть органических веществ имеет низкую теплопроводность, и для их анализа целесообразно использовать газы-носители с возможно более высокой теплопроводностью. Этому требованию удовлетворяют водород и гелий, но на практике водород ввиду его взрывоопасности применяется значительно реже гелия.
Ионизационно-пламенный детектор представляет собой камеру, в которой поддерживается водородное пламя, являющееся источником ионизации. В камеру вводят необходимые для поддержания пламени водород и воздух; водород в смеси с газом-носителем подается в детектор через канал горелки, а воздух - через другой канал и распределяется равномерно диффузором. Горелка является одним из электродов, она изолирована от корпуса и соединена с источником стабилизированного напряжения. Второй электрод, называемый часто коллектором, расположен над горелкой. Во внешнюю цепь включен электрометр, измеряющий ток между электродами.
Поскольку в пламени чистого водорода число ионов мало, сопротивление межэлектродного газового пространства очень велико (10 в 13 - 10 в 14 Ом) и ток детектора весьма мал (10 в -12 – 10 в -11 А). Этот ток, возникающий за счет ионизации примесей в газе-носителе, водороде и воздухе, является постоянным фоновым током детектора. При внесении с газом-носителем из колонки анализируемых органических веществ число ионов в пламени резко увеличивается, сопротивление пламени падает, и во внешней цепи детектора регистрируется соответствующее возрастание ионного тока.
Последовательность операций
Вначале устанавливают постоянную скорость подачи газа-носителя в хроматограф, нагревают детектор и колонку до определенной постоянной температуры, проверяют и настраивают цепь детектора. После этого вводят в колонку образец. В зависимости от концентрации и числа разделяемых компонентов объем вводимого газа-носителя колеблется от 1 до 10 мл, а объем жидкого образца - от 0,1 до 10 мкл. В системе ввода поддерживается такая температура, чтобы образец практически мгновенно испарялся.
Вместе с газом-носителем введенный образец в парообразном состоянии поступает в колонку. Колонка поддерживается в нагретом состоянии, но ее температура должна быть несколько ниже температуры кипения компонентов разделяемой смеси.
Отдельные компоненты образца в порядке их элюирования в колонке поступают в детектор, с помощью которого устанавливают наличие и количественное содержание каждого из них.
Метод газовой хроматографии не позволяет автоматически идентифицировать компоненты смеси по форме пика. Кривая элюирования показывает только число компонентов разделенной смеси.
Жидкостная хроматография
Жидкостная распределительная хроматография является аналогом газо-жидкостной. Разделение веществ также основано на различном их распределении между неподвижной и подвижной фазами.
В жидкостной распределительной хроматографии могут иметь место две системы фаз: неподвижная водная фаза (силикагель, на котором имеется слой воды) - подвижная органическая фаза; органическая неподвижная фаза - неорганическая подвижная фаза. В качестве неподвижной органической фазы используют гранулированные полимеры - полистирол, тефлон и другие материалы, способные сорбировать и удерживать на поверхности органические растворители. Используя такую систему, можно разделять как органические, так и неорганические вещества.
В жидкостной адсорбционной хроматографии в качестве неподвижной фазы обычно используют твердые активные адсорбенты с частицами малого размера: оксид алюминия, силикагель, цеолиты, целлюлозу, тальк и другие, а в качестве подвижной - органические растворители и их смеси.
Эффективность разделения зависит от различия в сродстве разделяемых компонентов к поверхности неподвижной фазы и элюенту.
Колоночная хроматография
Прежде чем перейти к описанию современных инструментальных методов жидкостной хроматографии, рассмотрим классическую схему разделения на хроматографической колонке.
Простейшие колонки изображены на рис. 178. Колонки заполняют гранулированным сорбентом таким образом, чтобы он образовывал столбик равномерной плотности. Для этого слой адсорбента механически уплотняют, или вносят адсорбент в виде однородной суспензии (из делительной воронки, снабженной эффективной мешалкой), или же вносят его в колонку, наполненную на 2/з объема растворителем. Во время загрузки растворитель должен каплями выливаться из нижнего конца колонки. Анализируемый раствор вводят в колонку и разделяют компоненты элюированием соответствующей подвижной фазы. Элюент через колонку внутренним диаметром 1 см пропускают со скоростью примерно 1 мл/мин. Фракции, содержащие обычно несколько миллилитров элюата, собирают вручную или с помощью автоматического коллектора фракций и определяют в них содержание веществ соответствующим аналитическим методом. Время выделения одного компонента составляет приблизительно 30 мин, т. е. для элюирования одного компонента требуется не менее 25 мл элюента.
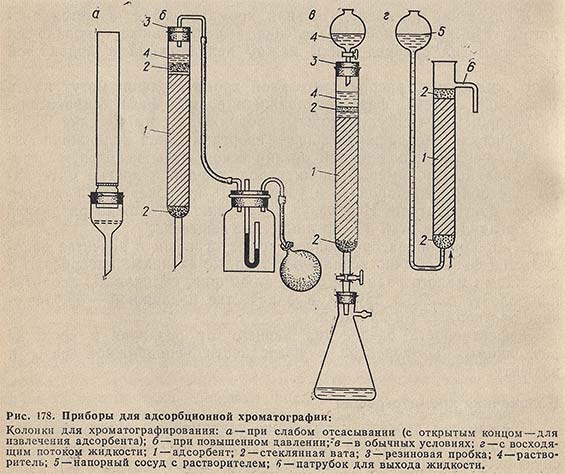
В современных жидкостных хроматографах элюент пропускают через колонку со скоростью в 100 раз выше, чем в обычной колоночной хроматографии.
При возрастании скорости элюента длина хроматографической колонки увеличивается, а для быстрого установления равновесия между неподвижной и подвижной фазами размер частиц твердого носителя должен быть очень мал.
Для пропускания элюента через плотно упакованную колонку с должной скоростью используют насос высокого давления. Объем раствора исследуемого образца составляет несколько микролитров, а масса образца измеряется в микрограммах. Образец вводят в верхнюю часть колонки с помощью шприца. Пройдя через колонку, элюент и растворенное в нем вещество попадают в детектор. Малые объем и масса образца позволяют получить узкие и четкие хроматографические пики.
На рис. 179 показана схема типичного колоночного хроматографа.
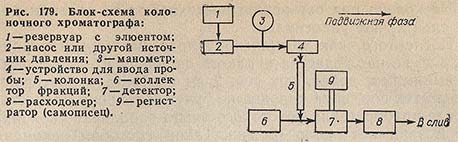
Элюент из резервуара 1 подается с помощью насоса 2 через устройство для ввода пробы 4 на вход колонки 5. Давление на входе в колонку контролируется манометром 3. Проходя через дозирующее устройство, элюент захватывает анализируемую пробу. Компоненты пробы выходят из колонки в виде разбавленного раствора в элюенте и поступают либо в коллектор фракций 6, либо в детектор 7 и затем в измеритель расхода 8. Сигнал детектора регистрируется на ленте самописца 9.
Существуют два основных способа ввода образца в колонку: с помощью шприца, когда образец наносят непосредственно в центр колонки на слой сорбента и более распространенный - с помощью крана-дозатора.
Длина и диаметр колонок влияют на степень разделения и способы их заполнения. Например, набивая колонки с внутренним диаметром 2 мм, трудно достичь хорошего уплотнения слоя. В то же время длиннее колонки, как правило, удобнее согнуть или свернуть кольцом. Это следует сделать до заполнения.
Прямые металлические колонки длиной до 1 м заполняют по следующей методике.
1. Закрепляют соединительное звено (адаптер) на нижнем конце колонки. Пористый фильтр будет удерживать сорбент.
2. Насыпают в колонку такое количество сухого сорбента, чтобы до уплотнения длина слоя была около 2 см.
3. Опускают в колонку металлический пруток или шток с тефлоновым наконечником. Шток должен быть плотно пригнан к колонке и вместе с тем свободно в ней двигаться.
4. Утрамбовывают слой набивки, осторожно постукивая нижней частью колонки о пол, так чтобы шток или трамбовочный пруток спрессовал слой неподвижной фазы.
5. Несколько раз осторожно постукивают по стенкам колонки, чтобы слой лучше уплотнился.
6. После того, как завершено уплотнение первой порции сорбента, т. е. не наблюдается никакого дальнейшего спрессовывания, шток вынимают, добавляют еще 2 см сорбента и повторяют операцию до тех пор, пока колонка не будет заполнена полностью.
После приобретения некоторого опыта заполнение колонки длиной 1 м занимает около 15 мин. Таким способом рекомендуется производить упаковку сорбента с размером частиц ^20 мкм. Для более тонких материалов эта методика непригодна; в настоящее время для них используются суспензионные методы.
В случае мягких (например, набухающих) сорбентов колонку заполняют 20-30% суспензией сорбента в элюенте, непрерывно прокачивая ее при малом давлении из сосуда, где обеспечивается эффективное перемешивание. Прокачивание осуществляют до установления постоянного давления; обычно это 20-30-кратный объем заполняемой колонки.
В случае жестких частиц сорбента, наоборот, 10-15% суспензию (в метиловом спирте, диоксане) прокачивают с максимально возможной скоростью (под максимальным давлением), иногда даже подают мгновенно - метод «впрыска» или «взрыва».
Инструментальные методы детектирования
Автоматические непрерывные коллекторы фракций (пробоотборники) карусельного или линейного типа обеспечивают отбор проб элюата. По мере заполнения приемника жидкостью, вытекающей из колонки до определенного объема или массы, автоматически подается следующий приемник. Однако необходимость в коллекторах фракций практически отпадает при наличии проточного чувствительного детектора.
Наибольшее распространение получили два детектора: дифференциальный рефрактометрический и спектрофотометрический. Рефрактометрический детектор обеспечивает непрерывную запись изменения показателя преломления элюата на выходе из колонки. Он наиболее универсален, так как практически всегда элюат и элюент имеют разные показатели преломления. При применении спектрофотометрического детектора измеряется поглощение элюатом падающего светового потока, длина волны которого может меняться от 200 до 700 нм.
Широкое распространение получили ультрафиолетовые фотометры, измеряющие поглощение на одной длине волны (обычно 254 нм), поскольку многие органические соединения содержат ароматические группировки и интенсивно поглощают лучистую энергию в этой области спектра.
В жидкостной хроматографии чрезвычайно важно непрерывно измерять скорость потока элюента, так как надежный качественный и количественный анализ затруднен, если скорость потока не постоянна.
В современных хроматографах используются, в основном, три типа измерителей потока. Счетчик капель регистрирует каждую вытекающую из колонки каплю, пролетающую между фотоэлементом и источником света. При наборе заданного числа капель на ленте самописца делается отметка, т. е. на хроматограмму как бы наносится масштаб объемного расхода элюента во времени. Пузырьковый отметчик времени измеряет время, за которое пузырек воздуха, введенный в поток элюента, проходит расстояние между двумя рисками в калиброванной стеклянной трубке, установленной на выходе из детектора. В сифонном расходомере используется объемный метод: подвижная фаза собирается в калиброванный сосуд до определенного объема (обычно 5 см3) и затем сосуд освобождается сифонированием. На ленте самописца автоматически отмечается каждый акт сифонирования.

В жидкостной хроматографии широко используется так называемое градиентное элюирование, т. е. непрерывное изменение состава элюента в процессе проявления хроматограммы. Так, если какой-то элюент один компонент вымывает очень быстро, а другие очень медленно, то, чтобы ускорить вымывание последних компонентов, желательно изменить состав элюента. Используя несложный прибор (рис. 180), позволяющий программировать состав элюента, можно смешивать два растворителя, постепенно меняя состав смеси таким образом, чтобы вначале преобладал один растворитель, а затем другой.
Последовательность операций
Навеску образца 0,05-5 мг растворяют в элюенте из расчета 0,5-5 мг/мл. Раствор должен быть оптически прозрачным и не содержать коллоидных взвесей. Удалить их можно фильтрованием через воронку с пористым фильтром с размером пор 5-10 мкм. Раствор не должен содержать избыточных количеств воздуха - для этого пробу вакуумируют с помощью водоструйного насоса. Хроматограф включают за 1-2 ч до начала работы и в процессе выхода его на рабочий режим проверяют наличие растворителя в сосудах для градиентного элюирования, отсутствие течей в жидкостных коммуникациях, исправную работу нагревателей. Критерием готовности прибора является ровная и устойчивая нулевая (базовая) линия на самописце при «холостом» протекании элюента через колонку и детектор. В момент ввода образца в колонку на ленте самописца делается отметка, которая служит началом отсчета при последующей идентификации разделенных компонентов.
Хроматография на бумаге
Бумажная хроматография является одним из видов жидкостной распределительной хроматографии.
В качестве носителя неподвижной жидкой фазы здесь применяется специальная фильтровальная бумага высокой степени чистоты и равномерной плотности. Считают, что неподвижной фазой служит постоянно присутствующая в целлюлозе вода. Подвижной фазой является органический растворитель или смесь растворителей.
Техника хроматографии на бумаге сводится к тому, что каплю раствора, содержащую 5-50 мкг исследуемой смеси веществ, наносят в виде точек на полоску или лист бумаги и высушивают горячим воздухом. Затем бумагу помещают в закрытый сосуд, в котором бумага непрерывно смачивается соответствующим растворителем - элюентом. Растворитель может двигаться в горизонтальной плоскости, подниматься снизу вверх (восходящая хроматография), сверху вниз (нисходящая хроматография) или движение растворителя может происходить от центра бумаги к периферии (круговая, или радиальная, хроматография).
Восходящая, нисходящая и круговая хроматография по технике выполнения может быть одномерной и двумерной. Для получения двумерных хроматограмм хроматографирование производят дважды во взаимно перпендикулярных направлениях: после проявления хроматограммы одним элюентом хроматограмму поворачивают на 90° и вторично проявляют другим элюентом.
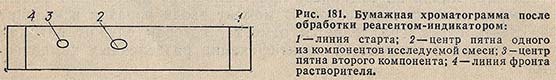
Для оценки хроматографического поведения веществ в определенных условиях эксперимента используют фактор Rf (от ratio of fronts - отношение фронтов), который равен отношению расстояния, пройденного веществом, к расстоянию, пройденному растворителем. Обычно для расчета Rf выбирают точку в центре пятна (рис. 181).
Абсолютные значения величин Rf трудно установить заранее, так как они изменяются в зависимости от условий проведения эксперимента. Однако они полезны для предварительной идентификации соединений, а при условии сравнения Rf исследуемого вещества и стандартного образца в одинаковых условиях достигается достоверная идентификация. Для этой цели на стартовую линию одной и той же бумажной полоски наносят три пробы: одну с исследуемым веществом, вторую со стандартным образцом и третью - со смесью равных количеств исследуемого и стандартного веществ. Количество хроматографируемого материала должно быть одинаковым во всех трех образцах. Если исследуемое вещество и стандартный образец идентичны по составу и строению, все три пятна после детектирования имеют одинаковую окраску и значения Rf; хроматограмма смеси в этом случае дает одно пятно.
Метод хроматографии на бумаге находит широкое применение для целей идентификации отдельных компонентов простых и сложных смесей, для качественного и полуколичественного их анализа, а также для установления степени чистоты препаратов.
Основные операции при хроматографировании на бумаге
Хроматографическое разделение веществ на бумаге состоит из следующих основных операций: подготовка бумаги и растворителей; нанесение пробы; хроматографирование и детектирование пятен на хроматограмме.
Бумага для хроматографических качественных анализов выпускается двух марок: «М» - бумага медленной впитываемости, № 1 и 2; «С» - бумага средней впитываемости, № 3 и 4. Бумага представляет собой 100% сульфатную целлюлозу, предгидролизную, кордную. Содержание воды около 6-7%. Обычная бумага гидрофильна. Однако в отдельных случаях, например при разделении смесей органических веществ, нерастворимых в воде, необходимо ее превратить в гидрофобную.
Для этого бумагу на 45 мин помещают в ацетилирующую смесь, состоящую из 90 мл уксусного ангидрида, 10 мл петролейного эфира и 8-10 капель конц. H2SO4. Затем ее вынимают и тщательно промывают в проточной воде в течение 15 мин, оставляют на 10-15 мин в дистиллированной воде, а затем промывают и высушивают в вакуум-эксикаторе над CaCl2.
Из бумаги вырезают соответствующего размера прямоугольные полосы или сворачивают из листа цилиндр, скрепляемый канцелярскими скрепками, для двумерной хроматографии.
Размеры полосок и цилиндра обусловливаются размерами камеры для хроматографирования, необходимой длиной перемещения разделяемых веществ и числом наносимых проб. Затем, отступя от нижнего края полосы бумаги, проводят простым карандашом линию старта, которая при хроматографировании должна быть на 20-30 мм выше уровня растворителя в камере для хроматографирования. На этой линии, на расстоянии 20-25 мм от краев бумаги, отмечаются кружками диаметром около 3 мм места, на которые наносят затем капля исследуемого раствора.
Для круговой хроматографии линией старта служит окружность диаметром 20-30 мм, проведенная из центра круга (см. рис. 183).
Затем бумагу выдерживают в камере для хроматографирования, на дно которой налита подвижная фаза (растворитель или смесь растворителей). Если в качестве неподвижной фазы применяют водные растворы нелетучих веществ (например, буферные растворы), то бумагу пропитывают этим раствором, высушивают, а перед применением выдерживают 10-16 ч в камере для хроматографирования.
Для пропитывания бумаги труднолетучими растворителями (формамид, пропиленгликоль и т. д.) их разбавляют метиловым спиртом, а затем бумагу погружают на 1-2 с в эту смесь, обжимают между листами фильтровальной бумаги и выдерживают 15-20 мин на воздухе.
Если бумага предназначена для хроматографирования аминокислот, ее вначале отмывают в 0,3 н. НСl, затем нейтрализуют 0,5 н. NH4OH, промывают дистиллированной водой до отрицательной реакции по фенолфталеину, пропитывают 0,1% фосфатным буферным раствором с pH = 7,0-7,5 и высушивают на воздухе.
Подготовка растворителей заключается в приготовлении смеси с взаимной насыщенностью подвижной и неподвижной фазы. Обычно для этого смесь, взятую в определенном соотношении, встряхивают в делительной воронке. После отстаивания органическую фазу наливают в лодочку (при нисходящей хроматографии) или в чашку (при восходящей). Водную фазу помещают в хроматографический сосуд, чтобы обеспечить насыщение атмосферы камеры парами обеих фаз.
Нанесение пробы на бумагу проводят до или после пропитывания ее неподвижной фазой. Каплю раствора (0,5-1 мг в 1 мл) наносят из капиллярной пипетки, легко прикасаясь ею к отмеченным местам на линии старта. Раствор должен заполнять только отмеченный кружок, и образующееся пятно должно иметь диаметр не более 5 мм. После высыхания пятна снова наносят каплю раствора и повторяют это несколько раз (после высыхания предыдущей капли) с таким расчетом, чтобы в целом нанести 0,01-0,05 мл раствора.
Затем бумажную полоску высушивают и применяют ее для нисходящей или восходящей хроматографии.
Одномерная восходящая хроматография
Осуществление восходящего варианта хроматографии не требует сложного оборудования, и исключена опасность перетекания растворителя. Как только фронт растворителя подойдет к верхнему концу бумаги, хроматографирование само собой прекращается.
Ближайший к линии старта конец бумаги с нанесенными пробами погружают на 20-30 мм в подвижную фазу, закрепляют бумагу и, закрыв камеру, дают растворителю медленно распространяться вдоль бумаги. В отдельных случаях, когда значения Rf компонентов смеси резко различаются, хроматографирование можно заканчивать, как только растворитель пройдет 100-150 мм от линии старта. Затем бумагу вынимают, карандашом отмечают на ней верхнюю границу фронта растворителя и подсушивают. Условия сушки зависят от летучести растворителя, свойств разделяемых веществ и реагента, применяемого для детектирования пятен.

На рис. 182 представлена камера для восходящей хроматографии. К пробке 2 цилиндра 1 прикреплен стеклянный стержень 3, служащий для подвешивания полоски хроматографической бумаги 4, вертикальное положение которой достигается с помощью зажимов 5. На дно цилиндра помещают подвижную фазу-растворитель 6. Пробка 2 герметизируется вазелином или консистентной смазкой.
Камеру, в которой происходит хроматографирование, плотно закрывают и устанавливают в термостат. Колебания температуры на время хроматографирования не должны превышать ±1,5°С.
Двумерная восходящая хроматография
Простое устройство для проведения восходящей двумерной хроматографии состоит из стеклянной кюветы, устанавливаемой под стеклянным колпаком на плоской подставке. В кювету с растворителем помещают цилиндр, сшитый из квадратного (200 X 200, 300 X 300 или 400 X 400 мм) листка хроматографической бумаги, с нанесенной и высушенной пробой. (Пробу наносят в нижнем левом углу квадрата в 50 мм от краев).
После того как фронт подвижной фазы достигнет верхнего края бумаги, хроматографирование прекращают, бумагу высушивают и поворачивают на 90° против часовой стрелки. При этом место нанесения капли исследуемого вещества окажется справа, а линия, по которой происходил подъем зон, - внизу.
В таком положении бумагу помещают в другую кювету с другой системой растворителей и снова хроматографируют. После достижения фронтом новой подвижной фазы верхнего края бумаги хроматографирование прекращают, бумагу высушивают и образовавшиеся зоны обнаруживают соответствующим реактивом.
Нисходящая хроматография применяется реже, и ее методическое оформление подобно восходящему варианту, только элюент движется сверху вниз.
Круговая (радиальная) хроматография
При круговой хроматографии вместо пятен на хроматограмме образуются концентрически расположенные кольца. Камерой для хроматографирования может служить небольшой эксикатор. Диаметр бумажного круга, в центр которого наносят пробу, должен на 20-30 мм превышать диаметр узкой части эксикатора. Насыщенный неподвижной фазой растворитель наливают на дно эксикатора. Бумажный круг, по радиусу которого вырезают полоску («фитиль») шириной 20 мм, помещают над узкой частью эксикатора, фитиль отгибают и опускают в растворитель (рис. 183). Скорость поступления растворителя к центру круга можно регулировать, изменяя ширину фитиля. Хроматографирование производят при герметически закрытом эксикаторе и, по возможности, при постоянной температуре. После того как растворитель дойдет почти до края бумажного круга, хроматографирование заканчивают, круг вынимают, высушивают и пятна детектируют. Круговую хроматографию на бумаге можно осуществить и в чашках Петри.
Детектирование пятен
Выбор метода детектирования зависит от химических свойств хроматографируемых веществ.
Расположение пятен бесцветных веществ на хроматограмме обнаруживают обычно при помощи реактивов, которые превращают разделенные компоненты смеси в окрашенные соединения. В ряде случаев положение вещества на хроматограмме можно обнаружить при рассматривании хроматограмм в затемненной комнате в ультрафиолетовом свете, под действием которого одни вещества начинают светиться, другие образуют темные пятна вследствие поглощения света.
После высушивания (на воздухе или в сушильном шкафу) хроматограмму опускают на несколько секунд в сосуд с раствором индикатора или опрыскивают им из пульверизатора. При этом бумажный цилиндр следует развернуть. Для. выявления окраски пятен хроматограмму после опрыскивания помещают на 5 мин в сушильный шкаф, нагретый до 90-100 °С, после чего пятна очерчивают карандашом.
В ряде случаев для сохранения окраски уже проявленную хроматограмму опрыскивают вторым реагентом или вводят соответствующее соединение в первоначальный раствор реагента-индикатора. Например, при детектировании пятен аминокислот нингидрином образуются пурпурные пятна, выцветающие при комнатной температуре в течение одного дня. Если в исходный нингидриновый реактив ввести ацетат кадмия или после детектирования нингидрином опрыскать пятна раствором ацетата никеле, окраска сохраняется пять лет.
В последние годы разработан и выпускается комплект аппаратуры для бумажной хроматографии, позволяющий автоматизировать нанесение пробы и хроматографирование при любой заданной температуре в интервале 10-40 °С. Специальное программное устройство позволяет начинать и заканчивать процесс хроматографирования в заранее заданное время.
Хроматография в тонком слое
Тонкослойная хроматография является одним из видов распределительной жидкостной хроматографии. Быстрота разделения смесей, относительная простота и универсальность обеспечили быстрое распространение этого метода, а использование гранулированного сорбента дает ему преимущества перед бумажной хроматографией из-за меньшего размывания пятен в направлении разделения.
Пластинки для хроматографирования
Разделение смесей производят на пластинках, покрытых тонким слоем адсорбента (силикагеля, оксида алюминия, полиамида, целлюлозы). Адсорбционный слой с пленкой растворителя, удерживаемого сорбентом, служит неподвижной фазой, а подвижной - индивидуальные растворители или смеси растворителей.
Техника хроматографирования в тонких слоях состоит в подготовке пластинок, нанесении пробы, проявлении хроматограммы и детектировании пятен. Положение пятна разделенного вещества описывается, так же как и в бумажной хроматографии, посредством измерения величины Rf, т. е. путем сопоставления с Rf стандартных веществ; вещества идентифицируются способом «свидетелей».
Тонкослойная хроматография применяется для идентификации веществ, определения чистоты вещества, качественного и количественного анализа веществ и их смесей, а в последнее время и для препаративных целей.
Существуют две модификации тонкослойной хроматографии: с закрепленным и с незакрепленным слоем адсорбента.
Тонкий слой адсорбента распределяют на подложке - пластинке из стекла, алюминиевой фольги или пластмассы. Чаще всего используют стеклянные пластинки (их легко очищать и можно использовать многократно) толщиной 2-4 мм, размером 3x8, 2,5x7,5; 5x15; 10x20; 20x20 см.
Пластинки промывают проточной водой, погружают на 24 ч в сосуд с 1% раствором моющего вещества, после чего тщательно промывают дистиллированной водой и высушивают, предохраняя от пыли. Брать пластинки следует за ребра, не касаясь поверхности. Хранить пластинки можно в эксикаторах или полиэтиленовых мешочках. Использованные пластинки для повторного применения сначала очищают от слоя адсорбента, после чего их промывают и высушивают, как указано выше.
Для приготовления пластинок с закрепленным слоем адсорбента применяют массу, состоящую из адсорбента, связующего материала и воды. В качестве связующего используют гипс CaSO4-1/2Н2О или крахмал. Иногда в состав массы для идентификации пятен разделяемых соединений добавляют флуоресцирующие вещества.
В случае применения гипса смесь из 5 г адсорбента (силикагеля или оксида алюминия), 0,2 г гипса и 12 мл воды растирают в фарфоровой ступке до однородной консистенции. Полученную массу наносят на подготовленную стеклянную пластинку и разравнивают при помощи специального устройства, обеспечивающего толщину слоя 0,15-0,25 мм по всей поверхности. В случае применения крахмала 28,5 г тонкоизмельченного и просеянного через сито № 0085 силикагеля, 1,5 г растворимого рисового крахмала и 54 мл дистиллированной воды нагревают при перемешивании до тех пор, пока раствор не начнет загустевать. Затем при постоянном перемешивании смесь охлаждают, размешивают с 20 мл дистиллированной воды и равномерно наносят на пластинки.
Пластинку с закрепленным слоем высушивают на воздухе в горизонтальном положении, после чего уже в вертикальном положении активируют нагреванием в течение 30 мин в сушильном шкафу при 110 °С и хранят в эксикаторе над СаCl2 или Mg(ClO4)2. В процессе сушки необходимо оберегать пластинки от загрязнения пылью или от поглощения паров летучих соединений, которые могут находиться в воздухе.
При нанесении тонкого слоя адсорбента на предметные стекла рекомендуется поступать следующим образом. Готовят суспензию из 44 г Аl2O3 и 1,6 г гипса в смеси 70 мл хлороформа и 30 мл метилового спирта или же из 30 г силикагеля и 1,5 г гипса в смеси 50 мл хлороформа и 50 мл метилового спирта. Суспензию тщательно перемешивают встряхиванием. Затем два предметных стекла, сложенных плоскостями, погружают в нее, медленно вынимают и дают стечь жидкости. После этого стекла разделяют, высушивают и хранят в эксикаторе над СаСl2. Если в поверхностном слое адсорбента появились трещины, пластинки непригодны к работе.
Очень удобны готовые пластинки «Силуфол» с тонким слоем Аl2O3 на подложке из алюминиевой фольги размером 150x150 мм и пластинки «Сата D-0», «Сата DS-0» с тонким слоем силикагеля, размером 100x100 и 200x200 мм, поставляемые странами СЭВ.
Пластинки с незакрепленным слоем подготавливают следующим образом. Адсорбент насыпают на стекло (лучше матовое) и разравнивают валиком, снимая при этом избыток. Валиком обычно служит стеклянная палочка диаметром 10 мм с надетыми на концы кусочками резиновой трубки. Расстояние между утолщенными концами трубки должно быть на 10-12 мм меньше ширины применяемой стеклянной пластинки. Толщину стенок трубки подбирают так, чтобы при накатывании образовался слой толщиной примерно 1 мм. Некоторые авторы рекомендуют распылять суспензию тонкоизмельченного адсорбента специальным распылителем по поверхности пластинки.
Пластинки с незакрепленным слоем приготавливают непосредственно перед употреблением.
Нанесение пробы и проявление хроматограммы
Раствор пробы наносят микропипеткой в 10-20 мм от края пластинки в виде точек или коротких черточек. В случае незакрепленного слоя необходимо следить, чтобы конец пипетки не касался слоя адсорбента.
В одну точку на пластинке можно наносить до 1 мг раствора пробы. Затем растворителю дают испариться и пластинки переносят в камеру для проявления. Пластинки с закрепленным слоем можно проявлять в наклонном или вертикальном положении; пластинки с незакрепленным слоем обычно ставят под углом 10-15°.
Камерой для проявления хроматограмм методом восходящей хроматографии может служить стеклянный сосуд с плоским дном, закрывающийся пришлифованной крышкой или стеклом. На дно камеры наливают столько растворителя, чтобы пластинка погружалась в него на 3-5 мм. Сначала помещают пластинку в слегка наклоненную камеру так, чтобы растворитель не смачивал стартовую полосу. Через 5-10 мин камеру возвращают в вертикальное положение. Для лучшего насыщения к стенке сосуда по всей высоте камеры прикрепляют смоченный растворителем лист фильтровальной бумаги, доходящий до дна сосуда.
При работе с закрепленным слоем адсорбента фронт продвижения растворителя на пластинке не должен превышать 100-150 мм. После того как фронт растворителя продвинется на 100-150 мм (на это требуется 15-90 мин), пластинку вынимают из камеры, отмечают карандашом линию фронта, высушивают и детектируют пятна, опрыскивая поверхность пластинки общими или специфическими реагентами.
В качестве общих реагентов для детектирования органических соединений используют конц. H2SO4, раствор Na2CrO4 в H2SO4, 1% раствор йода в метиловом спирте и др. Преимущество тонкослойной хроматографии перед бумажной состоит в том, что перечисленные общие реактивы не разрушают слой адсорбента, как разрушают бумагу. Весьма часто в качестве универсального реагента для детектирования пятен используют пары йода. Пластинку помещают в закрытую банку (эксикатор), содержащую несколько кристалликов йода. Пары йода растворяются в большинстве органических соединений, которые становятся заметными в виде коричневых пятен на бледно-желтом фоне. Пятна, обнаруженные с помощью йода, на воздухе быстро обесцвечиваются. Можно замедлить обесцвечивание и повысить чувствительность реакции, если проявленную пластинку опрыскать раствором 7,8-бензофлавона (0,3 г 7,8-бензофлавона в 95 мл этилового спирта + 5 мл 30% H2SO4). Этот реагент дает интенсивное голубое окрашивание с йодом.
Существует и другой универсальный способ детектирования разделенных веществ. При приготовлении пластинок для тонкослойной хроматографии к адсорбенту прибавляют неорганический фосфор и таким образом получают флуоресцирующие пластинки. Поскольку органические вещества чаще всего гасят флуоресценцию, то в ультрафиолетовом свете они обнаруживаются в виде темных пятен. Если вещества сами флуоресцируют, то под действием ультрафиолетового излучения они обнаруживаются в виде светящихся пятен на темном фоне.
Разработаны также методы количественного определения веществ в хроматографических пятнах непосредственно на пластинке. Для этих целей используются денситометры или флуориметры.
Комплект оборудования для тонкослойной хроматографии
Оборудование для тонкослойной хроматографии КТХ-01 отечественного производства предназначено для качественного анализа многокомпонентных смесей органических и неорганических веществ, а также для препаративного получения веществ, составляющих эти смеси. Оборудование поставляется в трех вариантах: в виде полного (КТХ-01-1), среднего (КТХ-01-2) или малого (КТХ-01-3) комплектов, различающихся наличием тех или иных устройств. В комплект КТХ-01 входит оборудование для приготовления хроматографических пластин, для нанесения пробы, для проведения тонкослойной хроматографии, для детектирования пятен химическими методами и для обнаружения их в ультрафиолетовом свете.
Адсорбенты для хроматографии
Адсорбенты, применяемые в хроматографии, кроме большой удельной поверхности, достигающей сотен квадратных метров на грамм, должны обладать избирательностью, химической й каталитической инертностью в отношении компонентов разделяемой смеси и подвижной фазы. Используемые в хроматографии адсорбенты должны быть, по возможности, монодисперсными (с одинаковым размером частиц), и состав их должен быть стандартным, что гарантирует воспроизводимость эксперимента.
Из адсорбентов, применяемых в хроматографии, назовем главнейшие: оксид алюминия, силикагели, силохромы, цеолиты и полисорбы.
Оксид алюминия - мелкокристаллический порошок амфотерного характера с удельной поверхностью 230-280 м2/г. Адсорбционные свойства Аl2O3 в сильной степени зависят от способа его приготовления, обработки и качества исходных материалов. Гигроскопичен. При увлажнении адсорбционная активность снижается. Оптимальной активности можно достигнуть нагреванием Аl2O3 при 400-450 °С в течение 5-6 ч. При хранении в отсутствие благи не теряет активности. Выпускается промышленностью: 1-й и 2-й активности (марки А-1 или А-2); по Брокману; оксид алюминия кислый, нейтральный и основной для тонкослойной хроматографии.
Силикагель - высушенный гель кремневой кислоты пористого строения с развитой внутренней поверхностью, кислого характера. Твердые хрупкие полупрозрачные или меловидные зерна от белого и желтоватого до коричневого цвета, с удельной поверхностью 300-700 м2/г - в зависимости от способа получения. Обладает резко выраженной гидрофильностью, в связи с чем широко применяется в качестве осушителя. Сорбирует пары многих органических соединений.
В зависимости от режима приготовления и формы зерен силикагели разделяются на кусковой - с зернами произвольной формы - и гранулированный. Промышленность выпускает большое число различных марок крупнопористых (КСК, ШСК, МСК, АСК) и мелкопористых (КСМ, ШСМ, МСМ, ACM) силикагелей, различающихся составом, пористостью и зернением. Особый интерес представляет силикагель марки ACM с радиусом пор 1,5 нм и величиной зерен 0,2-0,5 мм. Кроме силикагелей заводского изготовления, осуществляется полупромышленное производство. Специальный набор из семи марок силикагелей, по 0,25 кг каждого, выпускается для хроматографии.
Силохромы - зерна пористого диоксида кремния высокой химической чистоты. Они характеризуются весьма однородным распределением пор по размерам, высокой механической и термической устойчивостью. Применяются для разделения высококипящих неполярных и слабополярных веществ. Силохромы могут также быть использованы в качестве носителей для разделения сильно адсорбирующихся веществ. Выпускаются марки С-80 и С-120 с удельной поверхностью 60-100 и 100-140 м2/г соответственно.
Цеолиты (молекулярные сита) - мелкопористые гидратированные алюмосиликаты щелочных и щелочноземельных металлов, природные или синтетические. Наибольшее применение нашли синтетические цеолиты марок КА, NaA, СаА, СаХ, NaX (первые буквы означают преобладающий в цеолите катион, последняя - тип цеолита А или X). Поры цеолитов представляют собой сферические полости с диаметром 1,14 нм, соединенные друг с другом более узкими отверстиями (их называют окнами).
Для цеолита типа А в калиевой форме (марка КА) эффективный диаметр окон около 0,3 нм, у натриевой формы (марка NaA) - примерно 0,4 нм, а у кальциевой (СаА) - около 0,5 нм.
У цеолитов марки X эффективный диаметр окон значительно больше. Так, для СаХ он близок к 0,8 нм, а для NaX - около 0,9 нм. Таким образом, основное различие между цеолитами типов А и X заключается в размерах окон, соединяющих полости.
Особенностью адсорбционных свойств цеолитов является их молекулярно-ситовое действие. Адсорбироваться в первичной пористой структуре могут только те молекулы, диаметр которых меньше эффективного диаметра окон. Более крупные молекулы практически не адсорбируются. Таким образом, при адсорбции на цеолитах происходит как бы отсеивание более мелких молекул от более крупных. Емкость таких молекулярных сит значительно больше, чем емкость других адсорбентов.
Синтетические цеолиты общего назначения предназначаются для глубокой осушки и тонкой очистки газов и жидкостей, а также для разделения газообразных и жидких смесей.
Цеолиты КА, NaA, СаА, NaX, СаХ выпускаются в виде таблеток, гранул или шариков различных размеров. Мелкокристаллические цеолиты формуют, применяя до 15-20% связующих (обычно глин), после чего подвергают кратковременной (2-6 ч) термической обработке при 550-600 °С.
Промышленность выпускает набор синтетических цеолитов из четырех марок (NaA, NaX, СаА и СаХ) по 0,25 кг каждого.
Полисорбы - сополимеры стирола и дивинилбензола, используемые в хроматографии спиртов, жирных кислот, альдегидов, кетонов, нитрилов, а также для определения примеси воды в органических жидкостях.
Выпускаются полисорбы марок 4, 4Т, 10 и др.
Пористые стекла - пористый продукт, получаемый выщелачиванием боронатровой фазы из фазоразделенного натровоборосиликатного стекла. Отличаются однородностью размеров пор.
Для гель-хроматографии выпускается набор мелкозернистых стекол (МПС) с разными размерами пор: МПС-250 ГХ, МПС-750 ГХ, МПС-1150 ГХ, МПС-1600 ГХ и МПС-2000 ГХ. Цифры означают преобладающий диаметр пор (в ангстремах), а буквы ГХ - назначение (гель-хроматография).