Химические методы определения содержания углеводов
Определение содержания сахаров
Химические методы определения сахаров основаны на восстанавливающей способности моносахаридов и некоторых полисахаридов первого порядка, например мальтозы. Такие сахара содержат свободные альдегидные или кетонные группы и называются редуцирующими сахарами. Для определения тех сахаров, которые непосредственно восстанавливающей способностью не обладают, их предварительно подвергают гидролизу. Так, например, при определении сахарозы химическим методом ее подвергают инверсии и определяют количество полученного инвертного сахара.
Наиболее часто применяемые в контроле бродильных производств химические методы определения содержания сахаров можно разделить на две следующие группы:
1) методы, основанные на окислении сахаров щелочными растворами двухвалентной меди;
2) методы, основанные на окислении сахаров, содержащих свободную альдегидную группу (альдоз), - йодом в щелочной среде; сахара, содержащие свободные кетонные группы - кетозы, например фруктоза, в этих условиях не окисляются.
Методы, основанные на окислении сахаров щелочными растворами двухвалентной меди
Определение содержания сахаров по методу Бертрана. Для окисления сахаров применяют жидкость Фелинга, состоящую из двух растворов: Фелинг I и Фелинг II. Фелинг I представляет собой раствор сернокислой меди, Фелинг II - смесь раствора едкого натра и калийнатриевой соли винной кислоты (сеньетовой соли). Смесь растворов Фелинга I и Фелинга II неустойчива при хранении, поэтому эти растворы хранят отдельно и смешивают в равных количествах по объему в момент применения. Приготовить смесь заранее и хранить ее нельзя, так как гидрат окиси меди в щелочной среде медленно окисляет сеньетовую соль, причем выделяется закись меди.
При смешивании растворов Фелинга I и Фелинга II вначале образуется осадок гидрата окиси меди по уравнению
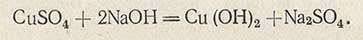
Осадок гидрата окиси меди сразу же растворяется, реагируя с сеньетовой солью с образованием комплексного соединения меди
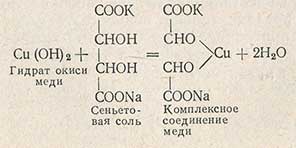
При взаимодействии комплексного соединения меди с редуцирующими сахарами при кипячении последние окисляются, а медь восстанавливается и выпадает в осадок красного цвета в виде закиси меди Сu2O.
Окисление сахаров жидкостью Фелинга является сложным процессом, приводящим к получению разнообразных продуктов распада и зависящим от многих условий, в том числе и от природы анализируемого сахара; следовательно, реакция окисления сахаров жидкостью Фелинга не может быть выражена стехиометрическим уравнением. Количество выпадающего осадка закиси меди зависит от следующих факторов: природы редуцирующего сахара; состава жидкости Фелинга, ее щелочности и общего объема нагреваемой жидкости; температуры и длительности нагревания; величины и формы сосуда, в котором ведется нагрев (кипячение); способа фильтрации и промывки осадка и пр.
Чтобы получить точные результаты, нужно применять растворы Фелинга определенного состава, кипятить в строго определенных условиях и соблюдать другие условия проведения определения. Тогда, пользуясь эмпирическими таблицами, можно по количеству выпавшего осадка закиси меди найти количество редуцирующего сахара.
Известно несколько десятков рецептов приготовления растворов Фелинга и проведения реакции окисления редуцирующих сахаров. В контроле бродильных производств применяют метод определения сахаров, разработанный Бертраном. Выпавший осадок закиси меди отфильтровывают под разрежением, промывают и добавляют к нему кислые железоаммиачные квасцы; при этом осадок одновалентной закиси меди растворяется и окисляется до двухвалентной меди, а железо восстанавливается из трехвалентного в двухвалентное:
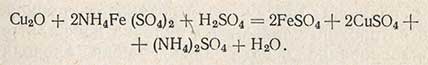
Полученный раствор титруют перманганатом калия; при этом двухвалентное железо окисляется в трехвалентное по уравнению
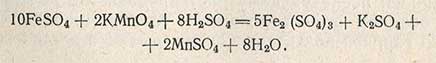
Ход определения по методу Бертрана следующий. В коническую колбу емкостью 100 мл отмеряют пипеткой 10-20 мл исследуемого раствора (с содержанием сахара не более 0,3-0,4%) и прибавляют по 10-20 мл растворов Фелинга I и II. Смесь нагревают до кипения в течение 3 мин, продолжая затем слабое кипячение точно 3 мин. По окончании кипячения колбу снимают с огня, дают образовавшемуся осадку 1-2 мин отстояться и фильтруют горячую жидкость через приготовленный асбестовый фильтр в трубке (рис. 41) под разрежением. Прибор для фильтрации (рис. 42) состоит из колбы Бунзена, в которую вставлена трубка с асбестовым фильтром или фарфоровым №2 или 3, предохранительной склянки и водоструйного насоса; колба и насос соединены между собой резиновой трубкой. Жидкость осторожно сливают по палочке на фильтр.
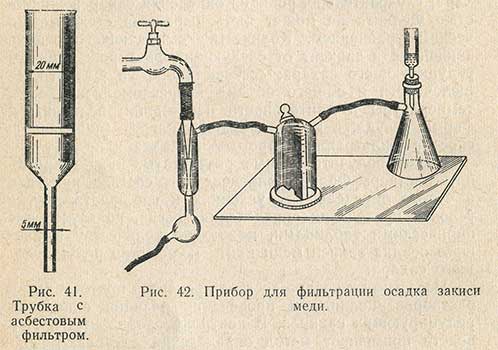
Фильтрование осадка закиси меди проводят при слабом разрежении. При фильтровании рекомендуется не переносить осадка закиси меди на фильтр, так как он образует на фильтре плотный слой, с трудом поддающийся последующему растворению. Когда жидкость над осадком отфильтрована, к осадку прибавляют немного горячей воды, дают снова осесть, а жидкость еливают через фильтр. При фильтровании нужно следить, чтобы осадок закиси меди в фильтре и в колбе все время оставался покрытым жидкостью во избежание окисления его кислородом воздуха.
Осадок промывают горячей водой до исчезновения щелочной реакции промывной воды. Затем фильтр снимают с колбы Бунзена, выливают из колбы собравшуюся там жидкость, ополаскивают колбу несколько раз водой и снова вставляют фильтр в колбу Бунзена. К осадку закиси меди в конической колбе приливают для растворения 25 мл раствора железоаммиачных квасцов, тщательно ополаскивают им стенки колбы и полученный раствор синевато-зеленого цвета сливают на фильтр. Для лучшего растворения осадка перешедшей на фильтр закиси меди верхний слой асбеста осторожно разрыхляют палочкой. Когда весь осадок растворится (на фильтре исчезнут черные крупинки окиси меди), включают насос и раствор переводят в колбу Бунзена. Коническую колбу, в которой проводилось кипячение, после этого тщательно промывают 5-6 раз холодной водой, каждый раз сливая ополоски на фильтр и отсасывая их. После этого прибор разбирают и фильтрат в колбе Бунзена титруют раствором перманганата калия до тех пор, пока зеленый цвет раствора от одной капли перманганата не перейдет в розовый.
При стоянии розовая окраска исчезает, так как окисляются не вполне отмытые органические вещества, но это во внимание не принимается. Для титрования применяют раствор перманганата, 1 мл которого соответствует 10 мг меди. Небольшое количество перманганата расходуется на окисление реактивов. Чтобы определить эту поправку на реактивы, параллельно проводят глухой опыт, в котором в отличие от основного вместо исследуемого раствора берут 10 мл воды.
Поправку выражают в миллилитрах раствора перманганата и при вычислении результатов вычитают ее из числа миллилитров перманганата, пошедшего на титрование сахарного раствора. По числу миллилитров перманганата, пошедшего на титрование (с учетом поправки на реактивы), находят количество меди, а затем, пользуясь таблицей Бертрана (см. табл. 6, 7, 8 приложения), по количеству найденной меди находят содержание в миллиграммах сахара во взятом объеме исследуемого раствора. Полученные данные можно пересчитать на 100 мл исследуемого раствора или 100 г продукта.
Определение содержания сахаров по методу Лейне и Эйнона. Существенным недостатком метода Бертрана является сложная фильтрация осадка закиси меди через асбестовый фильтр под разрежением, промывание осадка и растворение в растворе трехвалентного железа, что требует много времени. Более быстрым методом определения редуцирующих сахаров является метод Лейне и Эйнона, основанный на том, что редуцирующие сахара способны восстанавливать и при этом обесцвечивать - превращать в бесцветное лейкосоединение - метиленовую синь. Поэтому метиленовую синь можно использовать в качестве индикатора при окислении сахаров жидкостью Фелинга.
К строго определенному количеству жидкости Фелинга добавляют несколько капель раствора метиленовой сини и титруют исследуемым сахарным раствором. При титровании происходит восстановление двухвалентной меди в одновалентную; после того как вся окисная медь превратится в закисную, незначительный избыток редуцирующего сахара реагирует с метиленовой синью - обесцвечивает ее, благодаря чему резко изменяется окраска жидкости.
Необходимо учесть, что лейкосоединение под влиянием кислорода воздуха с течением времени вновь переходит в окрашенную метиленовую синь. При каждом определении проводят два титрования: предварительное и основное. При предварительном титровании в коническую колбу емкостью 150-200 мл пипеткой отмеряют по 10 мл растворов Фелинга I и II (приготовленных по рецепту Соксле) и нагревают до кипения. Затем, не прекращая кипячения, осторожно и постепенно приливают туда же из бюретки исследуемый сахарный раствор, все время взбалтывая колбу до тех пор, пока синий цвет кипящей смеси не исчезнет почти полностью. После этого прибавляют четыре капли метиленовой сини и, не прекращая кипячения, по каплям прибавляют раствор из бюретки до перехода синей окраски кипящей жидкости в красную или оранжевую от выпавшего осадка закиси меди. Продолжительность кипячения жидкости в колбе в течение всего титрования не должна превышать 3 мин (плитку разогревают раньше начала определения).

При титровании жидкости Фелинга исследуемым раствором следует брать коническую колбу с узким горлом, а для предохранения бюретки от нагрева к ее носику при помощи каучуковой трубки присоединяют тонкую стеклянную изогнутую трубку (рис. 43). Конец этой трубки, входящий в горло колбы, должен быть несколько оттянут. Лучше всего применять для титрования прибор, показанный на рис. 44. При втором (основном) титровании к смеси I и II растворов Фелинга в колбу прибавляют исследуемый сахарный раствор в количестве, на 0,5-1 мл меньшем, чем пошло на предварительное титрование. Смесь в колбе кипятят 2 мин и, не прекращая кипячения, добавляют 3-5 капель раствора метиленовой сини. Затем приливают из бюретки по 2-3 капли исследуемого раствора, давая смеси после каждого прибавления реагировать 2-3 сек до тех пор, пока синяя окраска не исчезнет и смесь не примет красной или оранжевой окраски.
Содержание сахара в граммах (х) в 100 мл исследуемой жидкости определяют по формуле
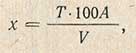
где Т - количество сахара, необходимое для восстановления 20 мл жидкости Фелинга (10 мл Фелинга 1 + 10 мл Фелинга II); при определении содержания гексоз Т = 0,0988 г; А - разведение исследуемой жидкости или навески исследуемого продукта; V - количество исследуемого раствора, пошедшее на титрование, мл.
Определение конца титрования по методу Лейне-Эйнона усложняется наличием красного осадка закиси меди, который мешает точно зафиксировать момент обесцвечивания метиленовой сини. Поэтому предложено добавлять к жидкости Фелинга раствор железистосинеродистого калия (желтой кровяной соли) K4Fe(CN)6. Осадок закиси меди Сu2O в момент образования реагирует с железистосинеродистым калием по уравнению
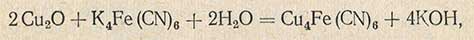
Образуя белый осадок, который не маскирует момента обесцвечивания метиленовой сини. Титрование заканчивают, когда синяя окраска исчезает и кипящая жидкость будет бледно-желтого цвета.
Определение содержания сахаров по методам Офнера и Мюллера. В методе Офнера окисление редуцирующих сахаров также проводят окисной солью меди, но в отличие от других методов предусматривается применение одного медьсодержащего раствора вместо двух растворов Фелинга. Окисление сахаров проводят реактивом Офнера, состоящим из сернокислой меди, сеньетовой соли, углекислого натрия и кислого двунатрийфосфата. В таком растворе концентрация гидроксильных ионов меньше, чем в жидкости Фелинга; поэтому раствор Офнера хорошо хранится и сеньетова соль незначительно восстанавливает двухвалентную медь. Полученный при взаимодействии редуцирующих сахаров и реактива Офнера осадок закиси меди окисляют титрованным (0,0323 н.) раствором йода, избыток йода оттитровывают 0,0323 н. раствором тиосульфата натрия (серноватистокислого натрия). По разности между количеством взятого 0,0323 н. раствора йода и количеством 0,0323 н. раствора тиосульфата натрия находят количество йода, вступившего в реакцию.
При определении редуцирующих веществ по методу Офнера содержание их не должно превышать 15 мг в 50 мл раствора или 7,5 мг в 25 мл раствора. Для определения берут 25-50 мл раствора, помещают в коническую колбу емкостью 300 мл и добавляют такое же количество (25 или 50 мл) реактива Офнера. Колбу ставят на асбестовую сетку с отверстием диаметром 6,5 см и нагревают до кипения в течение 4-5 мин, затем уменьшают пламя так, чтобы оно едва касалось сетки, и поддерживают умеренное кипение в течение 5-7 мин (при определении редуцирующих сахаров в белом сахаре 5 мин, в мелассе - 7 мин).
При нагреве на электрической плитке кипение регулируют, включая и выключая ее. После кипячения колбу охлаждают в холодной воде, не взбалтывая, во избежание окисления осадка. В охлажденную колбу вливают 7,5 мл 1 н. раствора уксусной или соляной кислоты, тотчас же прибавляют из бюретки 20 мл 0,0323 н. раствора йода (йод всегда должен быть в избытке), закрывают колбу стеклянной или корковой пробкой и, периодически перемешивая содержимое путем вращения, оставляют на 2 мин. Ровно через 2 мин оттитровывают избыток йода в колбе 0,0323 н. раствором тиосульфата натрия в присутствии 2.5 мл 0,5%-ного раствора крахмала. После прибавления крахмального раствора смесь окрашивается в темно-лиловый цвет, который при дальнейшем титровании постепенно переходит в синий, резко изменяющийся от одной капли в зеленый или бронзовый, это является признаком окончания титрования. Содержание редуцирующих сахаров вычисляют исходя из того, что 1 мл 0,0323 н. раствора йода соответствует 1 мг (0,001 г) гексоз.
При исследовании сахарсодержащих продуктов следует учитывать, что сахароза в небольшой степени окисляется реактивом Офнера. На каждый грамм сахарозы, содержащейся во взятой навеске, вводится поправка в 0,1 мл 0,0323 н. раствора йода. В некоторых сахарсодержащих продуктах, например мелассе, кроме редуцирующих сахаров, содержатся и другие вещества, окисляемые йодом. Для учета этих веществ проводят глухой опыт аналогично основному определению, но без кипячения. После внесения поправок на сахарозу и на вещества, окисляемые на холоду йодом, вычисляют содержание редуцирующих сахаров.
Можно также определять редуцирующие сахара по методу Мюллера, который в своих теоретических положениях подобен методу Офнера. Реактив Мюллера состоит из сернокислой меди, сеньетовой соли и углекислого натрия. При взаимодействии реактива Мюллера с редуцирующими сахарами выпадает осадок закиси меди. Полученный осадок окисляют - 1/30 н. раствором йода, избыток йода оттитровывают - 1/30 н. раствором тиосульфата натрия. Основное отличие метода Мюллера от метода Офнера состоит в том, что в методе Мюллера нагревание проводят в кипящей водяной бане, т.е. при постоянной температуре, исключающей перегрев.
Для определения по методу Мюллера 25-50 мл раствора помещают в коническую колбу емкостью 300 мл, добавляя разбавленную уксусную кислоту до нейтральной реакции по фенолфталеину, доводят водой до объема 100 мл, добавляют 10 мл раствора Мюллера и помещают колбу на 10 мин в кипящую водяную баню. Баня должна иметь такие размеры и вода в ней должна кипеть так, чтобы кипение не прерывалось при помещении в нее колбы. Колба должна быть настолько погружена в кипящую воду, чтобы находящийся в колбе раствор был на 2-3 см ниже уровня воды в бане. Колбу помещают на фарфоровую или металлическую подставку с круглыми вырезами так, чтобы она не касалась дна бани. После десятиминутного кипячения колбу вынимают из бани и, не взбалтывая, быстро охлаждают под струей холодной воды. Раствор должен иметь голубоватую или зеленоватую окраску.
Наличие желтой окраски раствора свидетельствует о недостаточном количестве реактива Мюллера. В этом случае опыт необходимо повторить с меньшим количеством исследуемого раствора. После охлаждения добавляют 5 мл приблизительно 5 н. уксусной или винной кислоты и 20-40 мл 1/30 н. раствора йода, смотря по количеству выпавшего осадка закиси меди, но так, чтобы йод всегда был в избытке. Затем колбу закрывают пробкой, содержимое колбы перемешивают вращательным движением и выдерживают 2 мин. После этого добавляют 5 мл 0,5%-ного раствора крахмала и оттитровывают избыток йода 1/30 н. раствором тиосульфата натрия.
Количество израсходованного тиосульфата натрия вычитают из прибавленного количества йода и таким образом находят количество йодного раствора, вступившего в реакцию. Из этого количества вычитают поправку на редуцирующую способность 10 мл раствора Мюллера. Эту поправку определяют так же, как и содержание сахара, но вместо исследуемого раствора берут 100 мл дистиллированной воды. Кроме того, как и в методе Офнера, вводят поправки на сахарозу и на вещества, окисляемые на холоде йодом. После внесения указанных поправок 1 мл израсходованного 1/30 н. раствора йода в условиях метода Мюллера соответствует 1 мг гексоз.
Методы, основанные на окислении альдоз йодом в щелочном растворе
Сахара, содержащие альдегидные группы (альдозы), в щелочном растворе количественно окисляются йодом в соответствующие одноосновные кислоты, а кетозы практически остаются без изменения. Этот метод определения альдоз разработан Вилынтеттером и Шудлем. Окисление глюкозы йодом в глюконовую кислоту протекает по уравнению
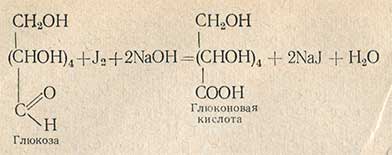
Для определения берут определенное количество титрованного раствора йода (йод должен быть в избытке), часть его расходуется на окисление, а избыток йода определяют титрованием тиосульфатом натрия; зная количество йода, израсходованное на указанную реакцию, можно вычислить количество глюкозы, окисленной в глюконовую кислоту. Реакция протекает стехиометрически в строго определенных условиях. Щелочность среды не должна превышать pH 9.
В сильно щелочных растворах окисление альдоз происходит не только с образованием одноосновных кислот, но и глубже, в результате чего расход йода получается завышенный. Однако количество щелочи должно быть достаточным для нейтрализации образующейся глюконовой кислоты. Кетозы в этих условиях не окисляются, но при значительном содержании их могут получаться завышенные результаты. Окисление кетоз происходит сильнее при повышенной температуре. Поэтому данное определение проводят при температуре 20° С.
Для определения в коническую колбу на 300 мл берут такую навеску вещества или такой объем раствора (с помощью пипетки), чтобы количество глюкозы в нем в сфере реакции не превышало 0,1 г; добавляют пипеткой 25 мл 0,1 н. раствора йода и 35-40 мл 1 н. раствора NaOH. Колбу закрывают пробкой и оставляют стоять в темном месте 15 мин; затем добавляют 5 мл 1 н. серной кислоты для нейтрализации щелочи и создания кислой среды.
Избыток йода оттитровывают тиосульфатом натрия в присутствии крахмала в качестве индикатора (крахмал добавляют в конце титрования) до исчезновения синей окраски. Одновременно ставят глухой опыт. Для этого в коническую колбу с помощью пипетки наливают 25 мл 0,1 н. раствора йода, приливают 1 н. раствор щелочи и ведут дальнейшую обработку и титрование тиосульфатом натрия в таких же условиях, как и при основном определении.
1 мл 0,1 н. раствора йода соответствует 0,009 г глюкозы. Содержание глюкозы в исследуемом продукте рассчитывают по формуле
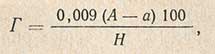
где Г - содержание глюкозы, %; A - число миллилитров 0,1 н. раствора тиосульфата натрия, пошедшее на глухой опыт; a - число миллилитров 0,1 и. раствора тиосульфата натрия, пошедшее на титрование исследуемой пробы; H - навеска вещества в сфере реакции, г.
Определение содержания крахмала
При определении химическим методом крахмал подвергают гидролизу до глюкозы, определяют количество полученной глюкозы, которое пересчитывают на крахмал. Гидролиз крахмала проводят в две фазы. В первой фазе крахмал осахаривают солодовой вытяжкой до мальтозы и декстринов:
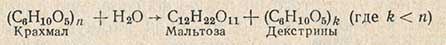
Во второй фазе гидролиз мальтозы и декстринов в глюкозу проводят под действием кислоты:
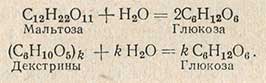
Суммарно реакцию гидролиза крахмала в глюкозу можно выразить уравнением
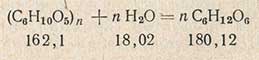
(приведенные цифры показывают молекулярную массу вещества). Одна часть глюкозы соответствует 162,1:180,12 = 0,9 части крахмала. Поэтому при пересчете глюкозы на крахмал количество глюкозы необходимо умножить на коэффициент 0,9.
Количество глюкозы определяют одним из ранее изложенных методов. Обычно пользуются методом Бертрана. Изложенный метод определения крахмала, разработанный Меркером, называют также химико-диастатическим методом, так как гидролиз крахмала проводят под действием фермента диастаза (старое название фермента амилазы).
Содержание крахмала в зерне определяют следующим образом. На аналитических весах взвешивают навеску 3,0000 г тонко размолотого зерна (помол должен проходить через сито с отверстиями диаметром 1 мм) и количественно переносят в мерную колбу на 200 мл. В колбу добавляют небольшое количество воды и тщательно размешивают в ней размолотое зерно до получения равномерной массы без комков, доливает воды примерно до половины колбы. Содержимое колбы тщательно перемешивают и погружают колбу в кипящую воду на 45 мин для клейстеризации крахмала. Для получения однородного клейстера в первые минуты нагревания суспензию в колбе энергично перемешивают. Затем содержимое колбы охлаждают до 63° С, добавляют 3 мл солодовой вытяжки и осахаривают в термостате при температуре 56-58° С.
Полноту осахаривания периодически проверяют пробой на йод. Для этого на белую фарфоровую пластинку выносят каплю пробы, содержащей жидкую и твердую фазы, охлаждают и смешивают с каплей 0,5%-ного йодного раствора. Появление синей или красной окраски свидетельствует, что осахаривание не закончено. Если несколько проверок покажет, что полнота осахаривания не достигнута, то добавляют еще 0,5 мл солодовой вытяжки и осахаривают до тех пор, пока при смешивании капли пробы с каплей йода получится желтая окраска. Чтобы избежать потерь, капли, взятые для йодной пробы, смывают обратно в колбу.
Осахаривание продолжают до исчезновения окрашивающихся йодом продуктов (крахмал, амило- и эритродекстрины). По окончании осахаривания содержимое колбы нагревают в кипящей водяной бане в течение 15 мин, охлаждают до 20° С и, удалив пену несколькими каплями серного эфира, доводят объем жидкости до метки. Содержимое колбы тщательно взбалтывают и фильтруют через сухой складчатый фильтр в сухую колбу. Первую порцию (20-25 мл) фильтрата отбрасывают. Фильтрат, содержащий мальтозу и декстрины, подвергают гидролизу разведенной соляной кислотой, причем эту операцию рекомендуется проводить тотчас же во избежание разложения глюкозы микроорганизмами.
Для гидролиза 50 мл фильтрата отбирают в мерную колбу на 100 мл, добавляют 4,5 мл 25%-ной соляной кислоты (относительная плотность 1,125), закрывают пробкой с воздушным холодильником (стеклянной трубкой длиной 50-60 см) и помещают на 2 ч в кипящую водяную баню. Во время нагревания следят, чтобы в бане не прекращалось энергичное кипение и уровень воды в ней был примерно на 1 см выше уровня жидкости в колбе, поэтому время от времени приливают в баню немного кипящей воды. Чтобы колба при сильном кипении не опрокинулась, горлышко ее вставляют в зажим (лапку) штатива. После двухчасового нагревания полученный раствор охлаждают, добавляют несколько капель метилоранжа и нейтрализуют до нейтральной или слабокислой реакции, постепенно и осторожно прибавляя 20%-ный раствор едкой щелочи. Нейтрализовать теплый раствор и допускать избыток едкой щелочи нельзя во избежание разложения глюкозы. После нейтрализации объем в мерной колбе доводят до метки дистиллированной водой и тщательно перемешивают содержимое.
В полученном нейтрализованном растворе определяют содержание глюкозы по методу Бертрана. Для этого в коническую колбу отмеряют пипеткой 10 мл нейтрализованного раствора, добавляют по 10 мл растворов Фелинга I и Фелинга II и далее ведут определение, как изложено ранее.
Пример. На титрование израсходовано 9,4 мл раствора перманганата с учетом поправки на реактивы. 1 мл раствора перманганата соответствует 10 мг меди. Содержание меди в исследуемой пробе 10*9,4 = 94 мг. По табл. 6 приложения находим, что 94 мг меди соответствует 49,22 мг глюкозы, откуда количество глюкозы во всей пробе
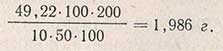
Крахмал осахаривают солодовой вытяжкой, содержащей определенное количество глюкозы. Для определения количества глюкозы отмеряют пипеткой 2 мл вытяжки в мерную колбу на 100 мл, добавляют 50 мл воды и 4,5 мл 25%-ной соляной кислоты, закрывают пробкой с воздушным холодильником и проводят гидролиз, как указано ранее, в течение 2 ч. Затем гидролизат охлаждают, добавляют несколько капель метилоранжа и нейтрализуют 20%-ным раствором едкой щелочи до нейтральной или слабокислой реакции. После нейтрализации добавляют воду до метки и тщательно перемешивают раствор. Из полученного нейтрализованного раствора отмеряют пипеткой 10 мл и определяют содержание глюкозы по Бертрану.
Пример. Допустим, что на титрование израсходовано 2,0 мл раствора перманганата (с учетом поправки на реактивы); это соответствует 20 мг меди, или 9,8 мг глюкозы. Во всей пробе раствора (100 мл) содержание глюкозы составит 98 мг. Указанное количество глюкозы содержится в 2 мл солодовой вытяжки; в 1 мл ее содержится 98:2 = 49 мг глюкозы. Для определения содержания крахмала взято 3 мл вытяжки, с которыми внесено
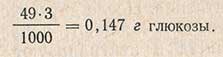
Крахмалистость зерна составит

где 0,9 - коэффициент пересчета глюкозы в крахмал; 3 - навеска зерна, г.
Найденное количество крахмала по существу представляет собой сумму содержания крахмала и пентозанов. Для точного определения крахмала следует найти содержание пентозанов и внести соответствующую поправку.
Определение содержания пентозанов
Пентозаны (С5Н8O4), являются составной частью гемицеллюлоз клеточных оболочек растений. В условиях химического метода определения крахмала пентозаны разлагаются с образованием пентоз - арабинозы и ксилозы (С5Н10О5):
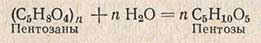
Пентозы содержат свободную альдегидную группу, окисляются жидкостью Фелинга, йодом и другими окислителями и искажают (завышают) результаты определения крахмала химическим методом. Определение содержания пентозанов основано на том, что при кипячении с кислотами происходит их гидролиз с образованием пентоз, а пентозы, теряя воду, превращаются в фурфурол:
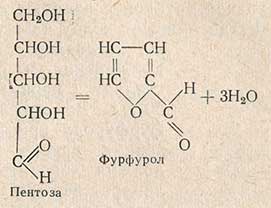
Глюкоза при перегонке с соляной кислотой разлагается с образованием оксиметилфурфурола. Последний дает те же реакции, что и фурфурол, и поэтому мешает его определению. Для устранения влияния оксиметилфурфурола следует проводить двойную перегонку. В отличие от фурфурола оксиметилфурфурол менее стоек и при нагревании разлагается. Количество фурфурола определяют бромным методом. Фурфурол в кислой среде присоединяет бром по уравнению
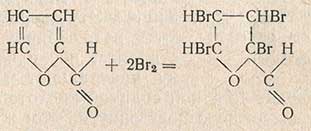
В качестве источника брома применяют смесь бромноватокалиевой соли и бромистого калия. Такая смесь при подкислении выделяет бром
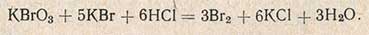
Избыток брома после реакции присоединения к фурфуролу определяют йодометрически
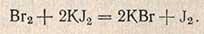
Выделившийся йод титруют тиосульфатом натрия. Определение содержания пентозанов в зерне проводят следующим образом. В круглодонную колбу помещают 100 мл фильтрата, полученного после осахаривания солодовой вытяжкой при химическом методе определения крахмала. В колбу добавляют 50 м 12%-ного раствора НСl. Колбу закрывают пробкой с капельной воронкой и отводной трубкой соединяют с холодильником (рис. 45). Устанавливают колбу на асбестовую сетку, нагревают содержимое ее до кипения и собирают дистиллят в мерный цилиндр на 50 мл. Во избежание потерь фурфурола при перегонке отводная трубка холодильника должна доходить до дна цилиндра, верхний конец которого закрывают ватой. После отгонки 30 мл жидкости в перегонную колбу через капельную воронку добавляют 30 мл 12%-ного раствора HCl, приливая его осторожно, чтобы жидкость в колбе не переставала кипеть.
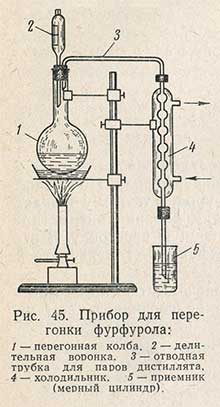
Одновременно дистиллят из приемного цилиндра переливают в мерную колбу на 300 мл. Эту операцию повторяют 9 раз, собирая 270 мл дистиллята, после чего перегонку прекращают и объем дистиллята доводят 12%-ной соляной кислотой до 300 мл. Затем проводят вторую перегонку на том же приборе и таким же образом, что и первую. Первые порции дистиллята (2-3 раза) возвращают в колбу для перегонки, так как за короткое время оксиметилфурфурол не успевает разложиться. В мерную колбу на 300 мл вновь собирают 270 мл дистиллята и доводят до метки 12%-ным раствором НСl.
Полученный раствор тщательно перемешивают. В коническую колбу на 200 мл переносят 100 мл дистиллята от второй перегонки и параллельно в такую же колбу помещают 100 мл 12%-ного раствора соляной кислоты (глухой опыт). В обе колбы прибавляют по 35 мл 0,1 н. бромной смеси (раствор КВгO3 и КВг) при определении пентозанов в овсе и ячмене или по 25 мл - при определении пентозанов в остальных культурах, плотно закрывают пробкой и ставят на 1 ч в темное место. Из бромной смеси в кислой среде выделяется бром, который присоединяется к фурфуролу. Через час в колбы добавляют по 10 мл раствора йодистого калия.
Избыток брома вытесняет йод, который тотчас же оттитровывают 0,1 н. раствором тиосульфата натрия в присутствии нескольких капель крахмала. Разница в количестве тиосульфата, пошедшего на глухой опыт и на титрование пробы, дает то число миллилитров 0,1 н. раствора брома, которое вступило в реакцию с фурфуролом. Содержание пентозанов в исследуемом зерне при навеске 3 г вычисляют по формуле

где П - содержание пентозанов, %; А - число миллилитров 0,1 н. раствора тиосульфата натрия, пошедшее на титрование выделившегося йода при глухом опыте; а - число миллилитров 0,1 н. раствора тиосульфата, пошедшее на титрование йода, выделившегося в исследуемом дистилляте; 0,0041 - количество пентозанов г, соответствующее 1 мл 0,1 н. раствора тиосульфата натрия.