Определение тяжелых металлов
Определение меди колориметрическим методом (арбитражный метод)
Метод заключается в минерализации испытуемой навески, отделении меди от других металлов при помощи сероводорода и последующего разделения сульфидов меди, цинка и свинца. Медь определяют колориметрически в виде комплексного медно-аммиачного соединения.
Методика определения. 15 г испытуемого продукта в фарфоровой чашке после высушивания (в случае необходимости) на водяной бане или в сушильном шкафу помещают в муфельную печь и осторожно при слабом накаливании сначала обугливают, а затем озоляют до белой или слегка серой золы. Озоление ведут очень осторожно при слабо-красном калении, так как при температуре выше 600° С соли меди могут улетучиваться. Для ускорения озоления можно к навеске прибавить смесь, состоящую из 0,2 г перекиси магния и уксуснокислого кальция или 0,5 г двууглекислого аммония с последующей обработкой золы 5-6%-ным раствором перекиси водорода. К золе прибавляют 5 мл соляной кислоты (1:1) и одну каплю пергидроля. Полученный раствор выпаривают на водяной бане досуха.
Сухой остаток в той же фарфоровой чашке обрабатывают 2 мл 10%-ного раствора соляной кислоты, добавляют 3 мл воды, размешивают стеклянной палочкой и фильтруют через бумажный фильтр, предварительно смоченный водой, в коническую колбу емкостью около 100 мл. Чашечку и фильтр промывают 15 мл дистиллированной воды, собирая промывные воды в ту же колбочку. Колбочку с раствором нагревают до температуры 40-50° С и через раствор в течение 40-60 мин пропускают сероводород. Выпавший осадок сульфидов и серы отделяют центрифугированием.
Жидкость сливают, осадок сульфидов 1-2 раза промывают 1%-ным раствором соляной кислоты, насыщенным сероводородом. К промытому осадку тотчас добавляют пять капель 10%-ного раствора едкого натра, нагревают в кипящей водяной бане, разбавляют 10 мл воды и центрифугируют. При большом осадке обработку раствором едкого натра повторяют 2 раза, жидкость не используют, а осадок в центрифужной пробирке, содержащей сульфиды свинца и меди, обрабатывают при осторожном нагревании 5-10 каплями смеси концентрированных серной и азотной кислот, взятых в равных количествах. Нагревание заканчивают после полного удаления паров азотной кислоты и появления белых тяжелых паров серного ангидрида (SO3). Пробирку охлаждают, добавляют по 0,5-1,0 мл дистиллированной воды и спирта. При наличии солей свинца в растворе появившуюся муть или выпавший белый осадок сернокислого свинца отделяют от находящейся в растворе меди центрифугированием. Раствор меди сливают в фарфоровую чашечку. Осадок сернокислого свинца промывают 2-3 раза 10 мл разбавленного этилового спирта (1:3 по объему), присоединяя промывную жидкость к раствору в фарфоровой чашечке. Раствор выпаривают досуха на водяной бане и после охлаждения прибавляют 1-5 капель 25%-ного раствора аммиака. При наличии в навеске следов меди (меньше 0,1 мг) раствор окрашивается в очень слабый голубой цвет. При более интенсивном окрашивании к раствору прибавляют 1-2 мл дистиллированной воды. В случае образования мути к раствору добавляют равный объем 25%-ного раствора аммиака и выпавший осадок отделяют центрифугированием.
Прозрачный раствор сливают с осадка в мерный цилиндр емкостью 10 мл. Осадок в пробирке 2 раза промывают небольшими количествами 1%-ного раствора аммиака. Промывные жидкости приливают к раствору в мерном цилиндре. Объем раствора в цилиндре доводят дистиллированной водой до 10 мл. Этот раствор используют для количественного определения меди. В пробирку для колориметрирования с делениями на 5; 10 и 15 мл переносят в зависимости от предполагаемого содержания меди часть или весь раствор. В три другие пробирки для колориметрирования наливают стандартный раствор меди, содержащий 0,1; 0,3 и 0,5 мг меди. Во все четыре пробирки добавляют по 2 мл 25%-ного аммиака и дистиллированной водой доводят объем растворов до 10 мл и хорошо перемешивают. Интенсивность окрашивания испытуемого раствора сравнивают с окраской типовых растворов. Если окраска испытуемого раствора интенсивнее окраски стандартного раствора, содержащего 0,1 мг меди, и слабее окраски раствора с содержанием 0,3 мг меди, считают, что в испытуемом растворе количество меди равно 0,2 мг.
Содержание меди (х) в миллиграммах на 1 кг продукта вычисляют по формуле
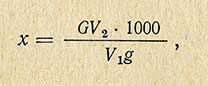
где G - количество меди, определенное при сравнении испытуемого раствора со стандартным, мг; V1 - количество исследуемого раствора, взятое для колориметрирования, мл; V2 - общее количество раствора, мл; g - навеска продукта, г.
Приготовление типового раствора меди
0,9821 г перекристаллизованной сернокислой меди (CuSO4-5Н2O) растворяют в небольшом количестве дистиллированной воды, переносят в мерную колбу емкостью 250 мл, добавляют 10 мл 10%-ной серной кислоты и доводят содержимое колбы дистиллированной водой до метки. 1 мл такого раствора соответствует 1 мг меди.
Определение меди йодометрическим методом (упрощенный стандартный метод)
Метод основан на переводе солей меди в солянокислые и последующем йодометрическом их определении. Реакция проходит следующим образом:
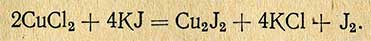
Выделившийся йод титруют гипосульфитом. Для маскирования солей железа и выведения их из реакции к раствору прибавляют однометаллический фосфат натрия, который образует с железом нерастворимое соединение, выпадающее в осадок и не реагирующее с йодистым калием:

Методика определения. В фарфоровую чашечку или в тигель берут навеску 50 г продукта, не содержащего поваренной соли, подсушивают (в случае необходимости) на песочной бане или в сушильном шкафу, а затем осторожно озоляют в муфельной печи при температуре не выше 600° С. Золу обрабатывают небольшим количеством разбавленной азотной кислоты (1:3 по объему) и выпаривают досуха на песочной бане. Затем к золе прибавляют 2-3 мл 10%-ной соляной кислоты и вновь выпаривают досуха. Остаток в чашке обрабатывают при нагревании 3 мл концентрированной уксусной кислоты с 10 мл дистиллированной воды и переносят без потерь в стакан небольшими порциями дистиллированной воды при помощи стеклянной палочки. Стакан и палочку промывают дистиллированной водой с таким расчетом, чтобы объем жидкости в стакане был не более 50 мл.
К раствору прибавляют 5 г однометаллического фосфата натрия. Полноту осаждения железа проверяют прибавлением к раствору 2-3 капель 10%-ного раствора роданистого аммония. При отсутствии розовой окраски к раствору прибавляют 3 г йодистого калия и выделившийся йод титруют при сильном встряхивании 0,01 н. раствором гипосульфита. Индикатором служит 1 мл 1 %-ного водного раствора крахмала. Для каждой серии определений проводят контрольное титрование всех применяемых реактивов без исследуемой навески. Реактивы берут в тех же количествах, что и при анализе испытуемого образца.
Содержание меди (в мг) на 1 кг продукта (х) вычисляют по формуле
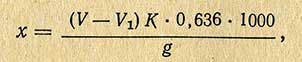
где V - количество раствора гипосульфита, пошедшее на титрование исследуемого образца, мл; V1 - количество раствора гипосульфита, пошедшего на титрование контрольного опыта, мл; g - навеска продукта, г; 0,636 - количество меди, соответствующее 1 мл точного 0,01 н. раствора гипосульфита, мг; К - поправка к 0,01 н. раствору гипосульфита.
Определение проводят в двукратной повторности.
Определение меди фотометрическим методом при помощи оксалилдигидразида (метод УкрНИИКПа)
В методе используется реакция между двухвалентной медью и оксалилдигидразидом в присутствии избытка ацетальдегида. Образующееся при этом комплексное соединение фиолетового цвета служит для количественного фотометрического определения содержания меди.
Методика определения. В фарфоровую чашку берут 10 г средней пробы продукта, прибавляют в качестве ускорителя 0,5 г двууглекислого аммония и озоляют в муфельной печи при температуре не выше 600° С. К золе прибавляют 3 мл раствора соляной кислоты (1:1), выпаривают досуха на песочной бане, после чего снова прибавляют 3 мл горячего 10%-ного раствора соляной кислоты и 3 мл горячей дистиллированной воды. Содержимое фарфоровой чашечки тщательно перемешивают стеклянной палочкой и количественно фильтруют через беззольный фильтр в мерную колбу емкостью 25 мл. Чашку и фильтр тщательно промывают небольшими порциями горячей дистиллированной воды, собирая их в ту же мерную колбу. Объем охлажденного раствора в колбе доводят дистиллированной водой до метки. После перемешивания раствор используют для проведения колориметрической реакции.
В пробирку, градуированную на 10 мл, переносят пипеткой 2,5 мл подготовленного раствора, прибавляют 1 мл 10%-ного раствора двузамещенного цитрата аммония, встряхивают и прибавляют концентрированный аммиак в количестве, предварительно установленном для нейтрализации реактивов. Затем в пробирку приливают 0,8 мл насыщенного водного раствора оксалилдигидразида, 2 мл 40%-ного раствора ацетальдегида и дистиллированной воды до объема 10 мл. Раствор перемешивают и проверяют pH при помощи индикаторной бумажки; pH должно быть 8,5-9. Через 10 мин измеряют оптическую плотность образовавшегося комплекса на фотоэлектроколориметре, применяя кювету толщиной слоя 10 мл и зеленый светофильтр. Вторую кювету фотоэлектроколориметра заполняют дистиллированной водой.
Калибровочный график строят по разбавленным растворам сернокислой меди, приготовленным из стандартного раствора этой соли, в следующих концентрациях: 0; 5; 10; 20; 30; 40; 50; 60 мкг меди в 10 мл. Стандартный раствор, содержащий 1 мг меди в 1 мл, приготовляют из 0,393 г перекристаллизованной CuSO4-5Н2O, растворенной дистиллированной водой в мерной колбе емкостью 100 мл. Растворы для составления калибровочного графика приготовляют следующим образом: стандартный раствор разбавляют в 100 раз, чтобы получить раствор, содержащий в 1 мл 10 мкг меди.
Содержание меди (в мг) в 1 кг продукта (х) вычисляют по формуле
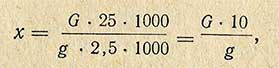
где G - количество меди в 2,5 мл раствора, найденное по графику; мкг; g - навеска, взятая для озоления, г; 25 - объем, до которого доведен раствор золы, мл; 2,5 - количество раствора, взятого для проведения колориметрической реакции, мл.
Определение проводят в двукратной повторности и среднюю арифметическую величину принимают за содержание меди. Одновременно с проведением колориметрирования исследуемого раствора измеряют оптическую плотность контрольного раствора, составленного из всех реактивов, применявшихся для проведения колориметрической реакции. Оптическую плотность контрольного опыта вычитают из оптической плотности испытуемого раствора.
Количество аммиака, необходимое для установления требуемого pH, определяют следующим образом: в пробирку вносят 2,5 мл испытуемого раствора, 2 мл 40%-ного раствора ацетальдегида и несколько капель 1%-ного раствора фенолфталеина. Пробирку встряхивают и к раствору прибавляют измеряемое количество концентрированного аммиака до появления ярко-малинового окрашивания, которое соответствует pH раствора 8,5-9,0.
Оксалилдигидразид приготовляют смешиванием эквимолекулярных количеств диметил- или диэтилоксалата и гидразингидрата, растворенных в пятикратных объемах этилового спирта. Выпавший в течение 1 я осадок оксалилдигидразида промывают небольшим количеством этилового спирта, перекристаллизовывают из кипящей воды и приготовляют его насыщенный водный раствор.
Определение железа колориметрическим методом
Метод основан на образовании окрашенного в красный цвет роданида железа (Fe(CNS)3) и его комплексных соединений (Fe(CNS6)---, Fe(CNS)++ при прибавлении раствора роданида аммония или калия к раствору, содержащему ионы трехвалентного железа.
Реакция проходит по уравнению
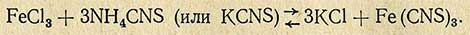
Fe(CNS)3, легко соединяясь с роданистым калием, образует комплексную соль железороданистого калия
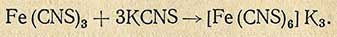
Методика определения. В фарфоровую чашечку взвешивают с точностью до 0,01 г 10 г средней пробы испытуемого продукта. Чашечку с навеской сначала помещают в сушильный шкаф для подсушивания (в случае необходимости), а затем в муфельную печь при небольшом нагреве. После прекращения выделения газообразных продуктов температуру в муфельной печи повышают до 600° С. Для ускорения озоления охлажденную золу смачивают несколькими каплями дистиллированной воды и 3%-ным раствором перекиси водорода. После подсушивания в сушильном шкафу золу снова помещают в муфельную печь для прокаливания. Смачивание и прокаливание золы продолжают до получения белой или слегка сероватой золы без частиц угля.
При значительном содержании в золе солей железа, марганца и меди она может быть слегка окрашенной. В охлажденную золу прибавляют 5 мл 2 н. раствора соляной кислоты и чашку помещают на кипящую водяную баню. Золу тщательно перемешивают стеклянной палочкой и после осаждения нерастворившихся частиц раствор осторожно, количественно по палочке переносят на гладкий фильтр и фильтруют в мерную колбочку емкостью 100 мл. Обработку золы в чашке соляной кислотой повторяют, фильтруя раствор через тот же фильтр. Затем чашку и фильтр 2-3 раза промывают горячей дистиллированной водой, подкисленной соляной кислотой. Промывные воды собирают в ту же мерную колбочку. После охлаждения раствор доводят дистиллированной водой до объема 100 мл. Этот раствор в количестве точно 10-25 мл (в зависимости от предполагаемого содержания в нем железа) выпаривают досуха в фарфоровой чашке на кипящей водяной бане.
Сухой остаток в чашке смачивают 2 мл концентрированной азотной кислоты и 2 мл воды и снова выпаривают досуха на водяной бане. Сухой остаток смачивают горячей дистиллированной водой, подкисленной соляной кислотой, тщательно перемешивают стеклянной палочкой и фильтруют в мерную колбочку емкостью 50 мл. Чашку и фильтр промывают 3 раза горячей подкисленной дистиллированной водой и промывные воды собирают в ту же мерную колбу. После охлаждения к раствору прибавляют несколько капель пергидроля, 1 мл концентрированной соляной кислоты и 2-3 мл 50%-ного роданистого калия или аммония, затем объем раствора в колбе доводят дистиллированной водой до метки. Окрашенный раствор в колбочке перемешивают, измеряют оптическую плотность на фотоэлектрическом колориметре, используя кюветы с толщиной слоя 10 мл и синий светофильтр. Параллельно проводят контрольный опыт на реактивы. Из величины оптической плотности испытуемого раствора вычитают величину оптической плотности контрольного опыта и по калибровочному графику находят соответствующее количество железа в миллиграммах.
Содержание железа в мг % (x) вычисляют по формуле
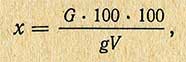
где G - количество железа, найденное по калибровочному графику, мг; g - навеска продукта, г; V - количество раствора, взятого для проведения цветной реакции, мл; 100 - общий объем вытяжки из золы, мл.
Определение проводят не менее чем в двукратной повторности и выводят среднеарифметическую величину.
Растворы для составления калибровочного графика приготовляют следующим образом: растворяют 0,8636 г железоаммиачных квасцов в дистиллированной воде, прибавляют 10 мл концентрированной серной кислоты (относительной плотностью 1,84) и объем в мерной колбе доводят до 1 л. В 1 мл этого раствора содержится 0,1 мг железа. Затем 10 мл этого основного стандартного раствора разбавляют в мерной колбе дистиллированной водой до 100 мл; 1 мл полученного рабочего раствора содержит 0,01 мг железа.
В мерные колбочки емкостью 50 мл вносят от 1 до 19 мл рабочего раствора, в каждую колбочку прибавляют несколько капель пергидроля, 1 мл концентрированной соляной кислоты и 2-3 мл 50%-ного раствора роданистого калия или аммония. Объем растворов в колбочках доводят дистиллированной водой до метки, растворы перемешивают и сейчас же измеряют их оптическую плотность. Одновременно проводят контрольный опыт с реактивом, оптическую плотность которого вычитают из оптической плотности соответствующих растворов.
Стандартные растворы для составления графика приготовляют не менее 3 раз и по средним арифметическим величинам их оптических плотностей строят график зависимости оптической плотности от количества железа в растворе. В указанных концентрациях калибровочный график представляет прямую линию.
Определение олова йодометрическим методом (объемный стандартный метод)
Метод основан на минерализации анализируемого продукта, извлечении соляной кислотой олова из золы с последующим восстановлением его водородом, образующимся при действии на алюминий соляной кислоты.
Реакция проходит по уравнению
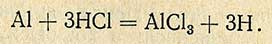
Выделившийся водород восстанавливает олово в двухвалентное
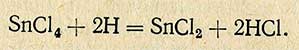
Двухвалентное олово количественно окисляется йодом по уравнению
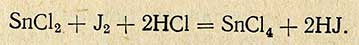
Количество йода, израсходованного на окисление олова, определяют титрованием гипосульфитом.
Методика определения. Минерализацию продукта можно проводить способом сухого озоления или окислением органических веществ концентрированными азотной и серной кислотами. Сухое озоление проводят следующим образом: навеску 25 г средней пробы измельченного и растертого продукта в фарфоровой чашке или тигле подсушивают (в случае необходимости) на кипящей водяной бане, осторожно обугливают на слабом огне или у открытой дверцы муфельной печи. После того как обугливание закончится и прекратится выделение газов, продолжают озоление при более высокой температуре, но не выше 600°С (темно-красное каление), так как при температуре выше 600° С олово может перейти в труднорастворимую метаоловянную кислоту. Озоление заканчивают, когда зола приобретет белый или серый цвет, без черных вкраплений. Озоление можно ускорить прибавлением к навеске 0,5 г двууглекислого аммония.
После охлаждения золу обрабатывают 10 мл соляной кислоты (1:1), хорошо перемешивают стеклянной палочкой, прикрывают фарфоровую чашечку часовым стеклом и нагревают на кипящей водяной бане в течение 15 мин. В чашку приливают несколько миллилитров горячей дистиллированной воды. Содержимое чашки хорошо перемешивают и фильтруют через маленький бумажный беззольный фильтр в коническую колбу емкостью 500 мл. Фарфоровую чашку, стеклянную палочку и фильтр тщательно промывают 3-4 раза горячей водой, каждый раз давая полностью профильтроваться раствору, находящемуся на фильтре. Промывные воды сливают в колбу с фильтратом. Общий объем должен составлять около 75 мл.
Фильтр с осадком помещают в тигель, подсушивают в сушильном шкафу, а затем осторожно озоляют. Золу количественно переносят в сухом виде из тигля через маленькую сухую воронку в колбу Кьельдаля емкостью 250 мл, прибавляют 25 мл концентрированной серной кислоты, смывая ею золу из воронки и со стенок горлышка колбы. Затем тигель, воронку и горлышко колбы ополаскивают несколькими миллилитрами дистиллированной воды. Колбу с золой и серной кислотой кипятят до полного растворения осадка. После охлаждения содержимое колбы количественно переносят в коническую колбу с солянокислым раствором золы. В колбу приливают 20 мл концентрированной соляной кислоты и воды до общего объема около 150 мл (отметка восковым карандашом или тушью на колбе). В этом растворе определяют содержание олова йодометрическим методом.
При проведении арбитражных анализов минерализацию проводят «мокрым» способом. В колбу Кьельдаля емкостью 500 мл через воронку с широким отверстием вносят 40 г хорошо растертой средней пробы анализируемого продукта и на кончике шпателя толченого химического стекла (стекло предварительно обрабатывают смесью равных объемов серной и азотной кислот). Остатки навески в фарфоровой чашке и на воронке смывают 50 мл 10%-ного раствора азотной кислоты. Колбу Кьельдаля с содержимым взбалтывают и оставляют в покое не менее чем на 10 мин (можно оставлять на ночь). Затем приливают 25 мл концентрированной серной кислоты (относительной плотностью 1,84), взбалтывают и в слегка наклонном положении ставят на сетку, в середине которой сделано небольшое круглое отверстие. В начале нагревания отверстие закрывают асбестом. Колбу закрепляют на штативе. Через капельную воронку с изогнутым концом, закрепленную на штативе таким образом, чтобы ее носик находился над центром колбы Кьельдаля, прибавляют к содержимому колбы Кьельдаля 150-200 мл концентрированной азотной кислоты. Приливают ее в процессе окисления постепенно по 15-20 капель в минуту. Содержимое колбы нагревают до кипения. При сжигании навески колба должна быть наполнена бурыми парами окислов азота. Если жидкость в колбе начинает темнеть, то увеличивают количество прибавляемой азотной кислоты до 30-35 капель в минуту. Когда жидкость в колбе станет бурой или бесцветной, количество прибавляемой азотной кислоты снова уменьшают до 15-20 капель в минуту. Через 20-30 мин, после окончания пенообразования, колбу нагревают на сильном огне, для чего асбест с отверстия сетки снимают.
При нагревании необходимо следить за тем, чтобы огонь охватывал дно колбы с жидкостью и не касался сухих стенок. После полного обесцвечивания и появления белых паров серного ангидрида жидкость кипятят еще 10 мин. Если жидкость остается бесцветной, то минерализацию считают законченной. В случае потемнения жидкости к ней снова прибавляют из воронки по каплям концентрированную азотную кислоту и продолжают кипячение, как указано выше.
После окончания минерализации бесцветную или слабо-зеленоватую жидкость охлаждают, добавляют в нее 25 мл насыщенного раствора щавелевокислого аммония и снова кипятят до выделения белых паров серного ангидрида. Охладив содержимое колбы, его переносят в коническую колбу емкостью 300 мл. Колбу Кьельдаля тщательно ополаскивают 60 мл дистиллированной воды и промывные воды присоединяют к раствору в конической колбе. Проточной водой охлаждают содержимое колбы, после чего добавляют 25 мл соляной кислоты (относительной плотностью 1,19). Этот раствор служит для йодометрического определения олова.
Коническую колбу с исследуемым раствором (после сухого озоления или «мокрой» минерализации) закрывают резиновой пробкой с двумя отверстиями. В одно отверстие вставляют трубку диаметром 5-6 мм, доходящую почти до дна колбы, для пропускания углекислого газа, в другое - такую же трубку, но заканчивающуюся под пробкой, для выхода углекислого газа.
Длинную трубку, которая проходит через промывную склянку с 5%-ным водным раствором сернокислой меди, соединяют с источником углекислоты - с баллоном или с аппаратом Киппа. Аппарат Киппа заряжают мрамором (углекислым кальцием) и соляной кислотой (1:1). В течение 5 мин через испытуемый раствор для удаления из него воздуха пропускают углекислый газ. Затем, не прекращая тока СO2, приоткрывают пробку в конической колбе, вносят в нее 0,4-0,5 г зерен алюминия или алюминиевой пыли и снова закрывают колбу пробкой, продолжая пропускать ток СO2. Через несколько минут, когда бурное выделение водорода ослабевает, колбу нагревают на асбестовой сетке над пламенем горелки или на электрической плитке так, чтобы жидкость в колбе не кипела. Когда весь алюминий растворится и останется только олово в виде губчатой массы, жидкость кипятят до полного растворения олова. Затем нагревание прекращают и усиливают ток углекислоты. Для охлаждения колбу с содержимым погружают в холодную воду. После охлаждения раствора прекращают пропускание СO2 и в колбу, приоткрыв немного пробку, вносят пипеткой 25 мл 0,01 н. раствора йода. Легким вращением колбы осторожно перемешивают содержимое. Стеклянные трубки обмывают дистиллированной водой. Воду сливают в ту же колбу. Общий объем в ней должен быть около 200 мл (отметка на внешней стороне стенки колбы). Полученный раствор титруют 0,01 н. раствором гипосульфита до соломенно-желтого цвета. Затем прибавляют 1 мл 1 %-ного раствора крахмала и продолжают титрование до обесцвечивания раствора. Параллельно проводят контрольный опыт с теми же количествами реактивов и в тех же условиях, но вместо испытуемого раствора берут дистиллированную воду.
Содержание олова (x) в миллиграммах на 1 кг исследуемого продукта вычисляют по формуле
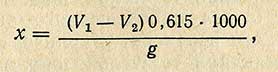
где V1 - количество 0,01 н. раствора гипосульфита, пошедшего на титрование 25 мл раствора йода, добавленного к контрольному опыту, мл; V2 - количество 0,01 н. гипосульфита, пошедшего на титрование 25 мл раствора йода, добавленного к исследуемому раствору, мл; g - навеска, взятая для анализа, г; 0,615 - (опытный коэффициент) количество олова, соответствующее 1 мл 0,01 н. раствора гипосульфита, мг.
Определение олова фотометрическим методом (метод УкрНИИКПа)
Метод основан на озолении испытуемого образца и на образовании комплексного соединения четырехвалентного олова с пирокатехиновым фиолетовым при pH 3,5 и последующем фотометрическом определении.
Методика определения. 5 г хорошо измельченной средней пробы продукта в фарфоровой чашке или в тигле озоляют с добавлением 0,5 г двууглекислого аммония до получения белой или слабо-серой золы. Золу обрабатывают в той же чашке 3 мл разбавленной соляной кислоты (1:3) и выпаривают на кипящей водяной бане досуха. Такую обработку проводят 2 раза. К остатку в чашке прибавляют 15 мл горячей (70-80° С) соляной кислоты (1:3), хорошо размешивают стеклянной палочкой. Раствор фильтруют через маленький беззольный фильтр в мерную колбу емкостью 50 мл. Фильтр с остатком промывают горячей водой, собирая ее в ту же мерную колбу. После охлаждения объем раствора в колбе доводят дистиллированной водой до метки (50 мл).
Перед фильтрованием определяют кислотность испытуемого раствора и выражают в миллиэквивалентах соляной кислоты. Для этого 1 мл испытуемого раствора титруют 0,1 н. раствором едкого натра в присутствии лакмуса. Затем в сухую пробирку отмеривают такое количество миллилитров 1 н. раствора соляной кислоты, чтобы общее содержание ее после добавления 100 исходного раствора составило 1 мэкв в 1 мл. Так, если в 1 мл раствора найдено 0,8 мэкв НСl, то в пробирку добавляют 0,2(1,0-0,8) мэкв НСl. Затем в пробирку к соляной кислоте прибавляют 2,5 мл 20%-ного раствора хлористого натрия и дистиллированной воды до объема 8 мл (отметка на пробирке), перемешивают, прибавляют 1 мл исследуемого раствора, две капли свежеприготовленного насыщенного раствора аскорбиновой кислоты или солянокислого гидроксиламина (для восстановления железа), снова перемешивают, прибавляют 0,5 мл 1%-ного раствора желатины, 0,5 мл 0,1%-ного спиртового раствора пирокатехинового фиолетового, 1 мл молярного раствора уксуснокислого натрия (для снижения pH раствора до 3,5). Раствор перемешивают и через 5 мин измеряют оптическую плотность раствора, окрашенного в зеленый цвет, по сравнению с контрольным раствором, в который внесены все реактивы, а вместо испытуемого раствора добавляют 1 мл дистиллированной воды. Окраска устойчива в течение 5 ч. Контрольная проба имеет желтую окраску.
Оптическую плотность раствора измеряют на фотоэлектроколориметре со светофильтром на 619 нм (миллимикрон) в кювете с толщиной слоя 10 мм. При проведении анализа следует строго придерживаться последовательности прибавления реактивов.
Содержание олова (x) в миллиграммах на 1 кг продукта вычисляют по формуле
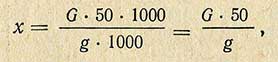
где G - количество олова в 1 мл раствора, найденное по калибровочному графику, мкг; g - навеска исследуемого продукта, г; 50 - разведение минерализованной навески.
Исходный стандартный раствор олова, содержащий 1 мг олова в 1 мл, приготовляют растворением 0,1 г металлического олова или 0,1901 г двухлористого олова (SnCl2-2H2O) в 30-40 мл соляной кислоты (1:3) при нагревании. Навески олова берут с точностью до 0,0001 г в небольшой стаканчик, при растворении олова стаканчик закрывают часовым стеклом. Полученный раствор охлаждают и для окисления двухвалентного олова в четырехвалентное в вытяжном шкафу к нему по каплям прибавляют бромную воду до появления слабого запаха или едва заметной желтой окраски. Избыток брома связывают 1-2 каплями 5%-ного раствора фенола. Раствор олова количественно переносят в мерную колбу емкостью 100 мл и доводят до метки водным раствором соляной кислоты (1:3). Из этого раствора приготовляют рабочий раствор, содержащий 10 мкг четырехвалентного олова в 1 мл, следующим образом:
в мерную колбу емкостью 100 мл вносят 25 мл 20%-ного раствора NaCl, 50 мл дистиллированной воды и 1 мл стандартного раствора олова, объем раствора в колбе доводят дистиллированной водой до метки.
Калибровочный график строят по растворам олова, приготовленным из рабочего раствора и содержащим от 0,5 до 40 мкг олова. Реакцию проводят так же, как и при определении испытуемого раствора.
В сухие пробирки приливают растворы реактивов в той же последовательности, что и при проведении определения в испытуемом растворе. В табл. 4 приведены количества соляной кислоты, поваренной соли, дистиллированной воды и рабочего раствора четырехвалентного олова, необходимые для приготовления растворов для составления калибровочного графика. Объем раствора во всех пробирках должен быть одинаковым.
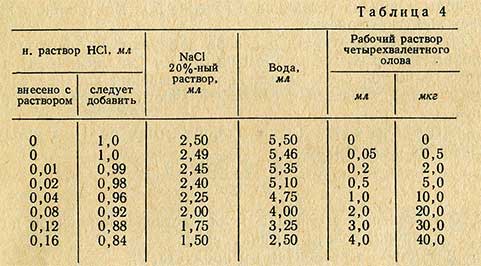
Величину измеренной оптической плотности растворов откладывают по оси ординат, концентрацию четырехвалентного олова - по оси абсцисс.
Определение свинца (стандартный метод)
Метод основан на отделении свинца (из раствора золы) от сопутствующих тяжелых металлов и нефелометрического его определения в виде хромата (РЬСгO4).
Реакция протекает по уравнению
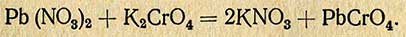
Методика определения. Навеску 15 г тонко измельченной средней пробы, подготовленной без применения металлических измельчителей, в фарфоровой чашке или тигле осторожно обугливают, а затем озоляют в муфельной печи при температуре не выше 500°С (слабо-красное каление).
При анализе жидких продуктов навески перед озолением выпаривают досуха, подсушивают на песочной бане, а затем осторожно озоляют. К белой или слегка серой золе прибавляют 5 мл соляной кислоты (1:1) и одну каплю пергидроля. Раствор выпаривают на песочной бане досуха. Сухой остаток в той же фарфоровой чашке обрабатывают 2 мл 10%-ной соляной кислоты, добавляют 3 мл дистиллированной воды и фильтруют через бумажный фильтр, предварительно смоченный дистиллированной водой, в небольшую коническую колбу. Фарфоровую чашку (или тигель) и фильтр промывают 15 мл дистиллированной воды, собирая промывные воды в ту же коническую колбу. В случае сплавления золы выщелачивание проводят 2 раза.
Полученный солянокислый раствор золы нагревают до 40-50° С и через оттянутую стеклянную трубочку, доходящую почти до дна колбы, пропускают в течение 40-60 мин сероводород. Сероводород получают в аппарате Киппа из сернистого железа и соляной кислоты (1:1). Сероводород пропускают через раствор в вытяжном шкафу.
Жидкость с выпавшим осадком сульфидов тяжелых металлов центрифугируют в пробирке емкостью 10 мл. После центрифугирования жидкость осторожно сливают с осадка при помощи маленького сифона. Осадок сульфидов в пробирке промывают 1-2 раза 1%-ным раствором соляной кислоты, насыщенной сероводородом. К промытому осадку сульфидов тотчас же добавляют пять капель 10%-ного раствора едкого натра и пробирку нагревают в кипящей водяной бане. Затем прибавляют 10 мл дистиллированной воды и осторожно перемешивают жидкость встряхиванием пробирки. Центрифугированием в этой же пробирке отделяют осадок сульфидов меди и свинца от растворившегося сульфида олова. При большом осадке сульфидов обработку раствором едкого натра повторяют.
В пробирку с осадком сульфидов меди и свинца приливают 5-10 капель смеси равных объемов крепкой серной и азотной кислот. Дно пробирки осторожно нагревают до полного удаления паров азотной кислоты и появления белых тяжелых паров серного ангидрида. Работу выполняют в вытяжном шкафу. К охлажденному раствору в пробирке прибавляют 0,5-1,0 мл дистиллированной воды и такое же количество этилового спирта. Если после прибавления воды и спирта раствор остается прозрачным, то соли свинца отсутствуют и проводить количественное определение свинца не надо. Появление в растворе мути или белого осадка сернокислого свинца указывает на наличие в растворе свинца и необходимость дальнейшего его количественного определения.
Осадок сернокислого свинца отделяют центрифугированием от растворившегося сульфида меди. Затем его промывают 2-3 раза небольшим количеством (около 10 мл) разбавленного спирта (1:1 по объему). К промытому осадку сернокислого свинца, оставшемуся в центрифужной пробирке, прибавляют 1 мл насыщенного раствора уксуснокислого натрия, предварительно слабо подкисленного уксусной кислотой, содержимое пробирки нагревают в кипящей водяной бане 5-10 мин, добавляют 1 мл дистиллированной воды, встряхивают пробирку и фильтруют через маленький бумажный фильтр, смоченный дистиллированной водой, в мерный цилиндр емкостью 10 мл. Пробирку и фильтр несколько раз промывают дистиллированной водой, собирая промывные воды в тот же цилиндр. Объем раствора в цилиндре доводят дистиллированной водой до 10 мл и хорошо перемешивают.
Для качественной проверки наличия свинца 5 мл раствора из цилиндра переносят в центрифужную пробирку, прибавляют три капли 5%-ного водного раствора двухромовокислого калия и хорошо перемешивают. Появление желтой мути от выпадающего хромата свинца (РЬСгO4) указывает на наличие свинца. Свинец считается необнаруженным, если раствор в течение 10 мин остается прозрачным.
Для количественного определения свинца 1 мл раствора из цилиндра переносят в плоскодонную пробирку с делениями емкостью 10 мл; в три другие такие же пробирки вносят типовой раствор азотнокислого свинца, содержащий 0; 0,1; 0,015 и 0,02 мг свинца. В пробирки с типовыми растворами азотнокислого свинца добавляют по 0,1 мл насыщенного раствора уксуснокислого натрия, слабо подкисленного уксусной кислотой. Затем во все четыре пробирки добавляют дистиллированную воду до 10 мл, перемешивают и прибавляют по три капли 5%-ного раствора хромовокислого калия. Растворы хорошо перемешивают и через 10-15 мин сравнивают с типовыми растворами. По степени помутнения испытуемый раствор и выбранный типовой должны быть одинаковыми.
Содержание уксуснокислого натрия в испытуемом и типовых растворах должно быть одинаковым, поэтому если для определения свинца берут не 1 мл, а какой-то другой объем, то в пробирки с типовыми растворами свинца следует добавлять столько уксуснокислого натрия, сколько его содержится в испытуемом растворе, взятом для определения.
Содержание свинца (х) в миллиграммах на 1 кг продукта вычисляют по формуле
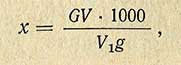
где G - количество свинца в типовом растворе, соответствующем по мутности испытуемому раствору, мг; V - количество испытуемого раствора, мл; V1 - количество испытуемого раствора, взятого для нефелометрического определения, мл; g - навеска продукта, г.
Приготовление типового раствора азотнокислого свинца. 160 мг азотнокислого свинца растворяют в дистиллированной воде в мерной колбе емкостью 100 мл, затем добавляют одну каплю концентрированной азотной кислоты, перемешивают и доводят содержимое дистиллированной водой до метки; в 1 мл этого раствора содержится 1 мг свинца. 2 мл приготовленного раствора переносят в другую мерную колбу емкостью 100 мл и доводят дистиллированной водой до метки; в 1 мл типового раствора содержится 0,02 мг свинца.