Определение витаминов
При глубоком изучении процессов пищеконцентратного и овощесушильного производства, при установлении пищевой ценности готовых продуктов, а также при контроле производства витаминизированных изделий определяют содержание в них следующих витаминов: витамина С (аскорбиновой кислоты), B1 (тиамина), B2 (рибофлавина), PP (никотиновой кислоты), каротина (провитамина A).
Подготовка проб при определении витаминов. Пробы исследуемых продуктов приготовляют непосредственно перед анализом. При анализе свежих плодов и овощей из отдельных экземпляров вырезают ножом из нержавеющей стали пробы в форме продольных сегментов, которые быстро измельчают ножом (капуста, лук) или на терке (картофель, корнеплоды), тщательно перемешивают и из полученной однородной массы отбирают пробу не менее 200 г, которую немедленно направляют на исследование.
Свежие ягоды и мелкие сочные плоды предварительно не измельчают; из средней пробы отбирают в банку из разных мест по нескольку ягод, плодов, перемешивают их и берут навеску для анализа. Из плодов и ягод с косточками удаляют косточки, а в дальнейшем поступают так, как описано выше.
Сухие плоды и овощи не менее 50 г измельчают на лабораторной мельнице или ножницами и полученный измельченный материал ссыпают в банку с притертой пробкой. Из тщательно перемешанной массы отбирают пробу для лабораторного анализа.
Пищевые концентраты в количестве не менее 200 г измельчают на лабораторной мельнице, перемешивают и отбирают пробу для анализа.
Витаминизированные молочные пищевые концентраты (в брикетированном виде) не менее 100 г измельчают и растирают в ступке, тщательно перемешивают и отбирают пробу для анализа.
Порошкообразные продукты в количестве не менее 50 г перед отбором пробы для исследования тщательно перемешивают.
При исследовании жидких, пюреобразных и пастообразных продуктов навески для анализа берут после тщательного перемешивания пробы.
Определение витамина C
Витамин C, l-аскорбиновая кислота (С6Н8O6), может находиться в пищевых продуктах в двух формах: восстановленной и окисленной (дегидроаскорбиновая кислота).
Количественные химические методы определения аскорбиновой кислоты основаны на ее восстановительных свойствах. Основными методами определения содержания аскорбиновой кислоты в препаратах и в пищевых продуктах является индофенольное или йодометрическое титрование. Применяемый индофенольный реактив - 2,6-дихлорфенолиндофенол, синего цвета, при титровании аскорбиновой кислоты восстанавливается и переходит в бесцветное лейкосоединение. Об окончании реакции судят по окрашиванию испытуемого раствора в розовый цвет, вызванному избытком индикатора, который в кислой среде имеет розовую окраску. По количеству индофенола, израсходованного на титрование, определяют содержание витамина С в продукте. При йодометрическом титровании применяют раствор йодноватокислого калия, индикатором служит крахмал.
При определении витамина C в пищевых продуктах применяют методы индофенольного титрования: арбитражный, с применением сероводорода и контрольный (упрощенный). Выбор метода зависит от свойств исследуемого продукта и назначения анализа.
Арбитражный метод (индофенольный с применением сероводорода)
Навеску исследуемого продукта 10-50 г в зависимости от предполагаемого содержания витамина C, взятую с точностью до 0,01 г, количественно при помощи 5%-ного раствора уксусной кислоты переносят в мерную колбу (или цилиндр) и этой же кислотой доводят содержимое колбы до объема 50-100 мл. При анализе концентратов и сушеных овощей и фруктов навеску 5-10 г растирают в ступке с 5-10 г стеклянного порошка или кварцевого песка (предварительно очищенного от примесей железа, промытого и прокаленного) и с трехкратным по отношению к навеске количеством 5%-ного раствора уксусной кислоты. При растирании анализируемый продукт должен быть полностью покрыт уксусной кислотой. Тщательно растертую смесь оставляют в ступке для настаивания на 10 мин, после чего содержимое ступки переливают в мерную колбу (или цилиндр) через воронку, стараясь не переносить осадка. Ступку, воронку и палочку несколько раз ополаскивают 5%-ным раствором уксусной кислоты, давая каждый раз отстояться осадку. Промывные жидкости сливают к испытуемому раствору в мерной колбе (или цилиндре) и доводят до объема 50-100 мл в зависимости от величины взятой навески и предполагаемого содержания витамина C. Содержимое мерной колбы или цилиндра тщательно перемешивают и центрифугируют или быстро фильтруют через слой ваты.
10 мл полученной уксуснокислой вытяжки пипеткой переносят в колбочку, стаканчик или центрифужную пробирку емкостью 60-80 мл и туда же прибавляют для создания необходимого pH и осветления раствора последовательно, при легком встряхивании, 0,4 г углекислого кальция и 5 мл 5%-ного раствора уксуснокислого свинца, приготовленного на 5%-ном растворе уксусной кислоты. Эту операцию следует проводить осторожно, так как прибавление углекислого кальция сопровождается пенообразованием. Раствор быстро центрифугируют или фильтруют в сухую колбочку через заранее приготовленный маленький складчатый фильтр.
Если фильтрат окажется мутным, то осветление повторяют на другой порции уксуснокислой вытяжки анализируемого продукта. Прибавляют к ней увеличенное в 2, 3 или 4 раза количество углекислого кальция и 5%-ного раствора уксуснокислого свинца, затем отфильтровывают или центрифугируют, как указано выше. Через прозрачный фильтрат в течение 5-15 мин пропускают ток сероводорода, получаемого из аппарата Киппа действием разведенной соляной (1:1) или серной (1:3) кислоты на сернистое железо. Для быстрого и полного осаждения сернистого свинца раствор в начале пропускания сероводорода энергично взбалтывают. Пропускание сероводорода заканчивают, когда слой жидкости над черным осадком сернистого свинца становится прозрачным. Раствор фильтруют через маленький сухой беззольный фильтр в сухую колбочку и из прозрачного фильтрата полностью удаляют сероводород током углекислого газа из баллона или аппарата Киппа, заряженного мрамором и разбавленной (1:1) соляной кислотой. Углекислый газ может быть заменен азотом. Контроль на полноту удаления сероводорода проводят при помощи фильтровальной бумаги, смоченной раствором уксуснокислого свинца, которую подносят к горлышку колбочки, в отсутствие сероводорода бумажка остается бесцветной, появление на ней желточерного пятна указывает на наличие сероводорода. Пропускание сероводорода и инертного газа следует проводить в вытяжном шкафу.
В колбочку предварительно приливают пипеткой 5 мл 80%-ного раствора уксусной кислоты и столько дистиллированной воды, чтобы общий объем жидкости с испытуемым раствором составил 15 мл. Затем вносят пипеткой от 1 до 10 мл испытуемого осветленного раствора, полученного после удаления сероводорода, и титруют из микробюретки или микропипетки 0,001 н. раствором 2,6-дихлорфенолиндофенола до появления розовой окраски, не исчезающей в течение 30-60 сек. Титрование проводят каплями при непрерывном легком встряхивании титруемого раствора. Титрование должно продолжаться не более 2 мин. После окончания титрования необходимо при энергичном взбалтывании раствора прибавить еще две капли раствора 2,6-дихлорфенолиндофенола; если окраска испытуемого раствора усилится, можно считать, что конец реакции был найден правильно, и в этом случае объем прибавленных капель индикатора не учитывают. При установлении количества испытуемого раствора, необходимого для титрования, следует исходить из того, чтобы на титрование израсходовалось не более 2 мл 0,001 н. раствора 2,6-дихлорфенолиндофенола.
Определение витамина C проводят не менее чем в двукратной повторности, причем результаты параллельных титрований не должны отличаться между собой более чем на 0,04 мл. Содержание витамина C вычисляют как среднюю арифметическую величину из 2-3 параллельных определений. При вычислении результатов титрования следует вводить поправку на контрольное определение: титрование 0,001 н. раствором 2,6-дихлорфенолиндофенола смеси 5 мл 80%-пой уксусной кислоты и 10 мл дистиллированной воды до появления розового окрашивания. Эту поправку, равную обычно для объема 15 мл 0,06-0,08 мл, вычитают из общего количества индикатора, пошедшего на титрование испытуемого раствора.
Содержание витамина C в продукте (х), выраженное в миллиграммах на 100 г, вычисляют по формуле
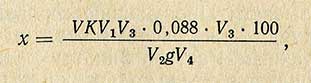
где V - количество 0,001 н. раствора 2,6-дихлорфенолиндофенола, пошедшего на титрование с учетом поправки на контрольное титрование, мл; К - коэффициент пересчета на точно 0,001 н. раствор 2,6-дихлорфенолиндофенола; V1 - объем, до которого доведена навеска при прибавлении к ней экстрагирующей жидкости, мл; V2 - объем анализируемой жидкости, взятой для титрования, мл; V3 - объем первоначального раствора или экстракта, взятого для анализа после прибавления уксуснокислого свинца, мл; V4 - объем первоначального раствора или экстракта, взятого для анализа перед обработкой уксуснокислым свинцом; g - навеска продукта, г; 0,088 - количество аскорбиновой кислоты, соответствующее 1 мл точно 0,001 н. раствора 2,6-дихлорфенолиндофенола.
Определение витамина C следует проводить не на прямом солнечном свете. Продолжительность анализа должна быть не более 1 ч.
Приготовление 0,001 н. раствора индикатора 2,6-дихлорфенолиндофенола
0,25-0,3 г индикатора взбалтывают в однолитровой мерной колбе с 600 мл дистиллированной воды в течение 1,5-2 ч (можно оставлять для растворения на ночь), доливают дистиллированной водой до 1 л, хорошо смешивают и фильтруют. Раствор индикатора пригоден для анализа в течение 5-10 дней. Хранить его следует в темноте, в прохладном месте, желательно в холодильнике.
Титр индикатора проверяют ежедневно. Появление при проверке титра грязноватого оттенка указывает на непригодность раствора индикатора для анализа.
Определение титра раствора индикатора - 2,6-дихлорфенолиндофенола
Титр раствора индикатора можно установить двумя способами.
Первый способ. К 5 мл раствора индикатора прибавляют 2,5 мл насыщенного раствора щавелевокислого натрия и титруют из микробюретки 0,01 н. раствором соли Мора, приготовленным на 0,02 н. растворе серной кислоты, до исчезновения синей окраски и перехода синевато-зеленоватого цвета в янтарно-желтый. Титр раствора соли Мора устанавливают по 0,01 н. раствору марганцовокислого калия, а титр последнего - по 0,01 н. раствору щавелевокислого натрия или щавелевой кислоте по общепринятым методикам.
Раствор соли Мора остается пригодным для анализа в течение 2-3 месяцев при хранении его в темном прохладном месте. Титр раствора соли Мора проверяют не реже 1 раза в месяц.
Второй способ. Несколько кристалликов аскорбиновой кислоты (примерно 1-1,5 мг) растворяют в 50 мл 2%-ного раствора серной кислоты. 5 мл этого раствора, взятого пипеткой, титруют раствором 2,6-дихлорфенолиндофенола из микробюретки до появления розового окрашивания, не исчезающего в течение 3 мин. Параллельно такой же объем (5 мл) раствора аскорбиновой кислоты титруют из другой микробюретки точно 0,001 н. раствором йодноватокислого калия (0,3568 г KJO3, высушенного в течение 2 ч при 105° С, растворяют в 1 л дистиллированной воды, полученный 0,01 н. раствор KJO3 перед анализом разбавляют в мерной колбе дистиллированной водой в 10 раз). Титрование проводят в присутствии нескольких кристалликов (1-2 мг) йодистого калия и 2-3 капель 1%-ного раствора крахмала до появления голубого окрашивания. Это титрование удобно проводить в фарфоровой чашечке.
Титр раствора 2,6-дихлорфенолиндофенола (х) по аскорбиновой кислоте вычисляют по формуле
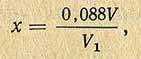
где V - количество 0,001 н. раствора KJO3, пошедшего на титрование раствора аскорбиновой кислоты, мл; V1 - количество раствора 2,6-дихлорфенолиндофенола, пошедшего на титрование раствора аскорбиновой кислоты, мл; 0,088 - количество аскорбиновой кислоты, соответствующей 1 мл точно 0,001 н. раствора 2,6-дихлорфенолиндофенола, мг.
Контрольный упрощенный метод определения витамина С
Метод применяется при массовых анализах свежих плодов и овощей. Он позволяет определять аскорбиновую кислоту только в восстановленной форме. Точность метода ±20%.
Методика определения. Во взвешенный стаканчик берут в зависимости от предполагаемого содержания витамина C в продукте навеску 10-30 г и быстро заливают ее 50 мл 4%-ного раствора соляной кислоты; навески, залитые кислотой, можно хранить в течение 10-15 мин. Навеску вместе с кислотой переносят в фарфоровую ступку. Часть кислоты из ступки сливают в мерную колбу или цилиндр емкостью 100 мл, а навеску с небольшим количеством оставшейся кислоты тщательно растирают. Затем содержимое ступки переносят в тот же цилиндр (или колбу), в котором находится остаток соляной кислоты, смывая дистиллированной водой остаток из фарфоровой ступки в ту же мерную колбу (или цилиндр). Раствор в мерной колбе доводят дистиллированной водой до метки. Содержимое колбы хорошо перемешивают и быстро фильтруют через марлю или воду. Из этого раствора отбирают пробу для титрования.
В случае труднорастираемых продуктов к навеске в фарфоровой ступке прибавляют 2-5 г взвешенного, хорошо промытого и прокаленного кварцевого песка или стеклянной пудры. После того как все содержимое ступки перенесено в мерную колбу (или цилиндр) и объем вытяжки доведен до 100 мл, к вытяжке добавляют дистиллированную воду в количестве 0,35 мл на каждый грамм взятого песка и всю жидкость снова хорошо перемешивают.
При исследовании жидкого материала его разбавляют в цилиндре 4%-ным раствором соляной кислоты и дистиллированной водой с таким расчетом, чтобы конечная концентрация соляной кислоты составляла 2%. Соляная кислота может быть заменена метафосфорной или щавелевой кислотой. Для получения вытяжки пользуются 2%-ным раствором метафосфорной кислоты, приготовленной на 2 н. растворе серной кислоты. Сначала приготовляют 20%-ный раствор метафосфорной кислоты на 2 н. растворе серной кислоты, а перед употреблением этот раствор разбавляют в 10 раз 2 н. раствором серной кислоты.
Навеску исследуемого продукта растирают в ступке с 2%-ным раствором метафосфорной кислоты (навеска должна быть покрыта кислотой), затем ее переносят в мерный цилиндр. Ступку несколько раз промывают небольшим количеством раствора метафосфорной кислоты, сливают эти растворы в цилиндр, доводя содержимое до 100 мл. Витамин C в растворе метафосфорной кислоты стабилен в течение нескольких часов. При отсутствии метафосфорной кислоты можно пользоваться щавелевой кислотой. Навеску исследуемого материала быстро растирают в ступке под 20 мл 1%-ного раствора соляной кислоты и затем переносят содержимое фарфоровой ступки в мерный цилиндр емкостью 100 мл и доводят объем вытяжки до 100 мл при помощи 1%-ного раствора щавелевой кислоты. После перемешивания вытяжку фильтруют. Для титрования 0,001 н. раствором 2,6-дихлорфенолиндофенола отбирают от фильтрованной вытяжки не более 5 мл.
Титрование и вычисление содержания витамина C (в миллиграммах на 100 г продукта) производят так же, как и в арбитражном методе. Расхождение между результатами анализов двух параллельных навесок из одного продукта не должно превышать 3-4%.
Метод определения витамина C в сульфитированных сушеных продуктах
Метод основан на том, что сернистые соединения (в кислой среде) блокируются формальдегидом и не мешают титрованию аскорбиновой кислоты.
Навеску сушеного продукта, взятого с таким расчетом, чтобы витамина C в вытяжке содержалось 0,04-0,1 мг, растирают в ступке с 5%-ным раствором метафосфорной кислоты. Вытяжку фильтруют и в случае исследования несульфитированного продукта титруют 0,001 н. раствором 2,6-дихлорфенолиндофенола.
При анализе сульфитированного сушеного продукта полученную метафосфорную вытяжку подкисляют 50%-ным раствором серной кислоты и обрабатывают формальдегидом, концентрация которого в конечном растворе должна быть 4%. Раствор оставляют стоять на 8 мин, а затем титруют 0,001 н. раствором 2,6-дихлорфенолиндофенола, как указано выше.
Определение каротина
Методы определения каротина основаны на извлечении его из растительных тканей бензином или петролейным эфиром и последующем освобождении от сопутствующих веществ при помощи адсорбционной хроматографии. Количественное определение каротина проводят колориметрированием полученных растворов, содержащих каротин. Для определения каротина предложены три варианта метода.
Методика определения. Первый вариант. Каротин извлекают из растительного материала после обезвоживания его спиртом или ацетоном, а затем омыляют вещества, перешедшие в экстракт, спиртовым раствором щелочи. Повторно извлекают каротин, фильтрат пропускают через адсорбционную колонку и затем определяют интенсивность окраски фильтрата.
Навеску измельченного продукта берут в количестве от 1 до 50 г в зависимости от содержания каротина и растирают ее в фарфоровой ступке с небольшим количеством промытого и прокаленного песка или измельченного стекла. К растертой массе в ступку приливают спирта или ацетона пятикратное количество, растирают, а затем добавляют порциями 20-30 мл бензина или петролейного эфира. Смесь растирают, экстракт фильтруют через бумажный фильтр; экстрагирование повторяют до тех пор, пока последние порции экстракта не станут бесцветными.
Фильтрат переносят в делительную воронку, добавляют несколько миллилитров дистиллированной воды для разделения слоев: верхний - бензиновый, нижний - спиртовой или ацетоновый. В другую делительную воронку сливают спиртовой или ацетоновый слой и промывают 2 раза бензином или петролейным эфиром, присоединяя эти вытяжки к основному фильтрату. Соединенные вытяжки переносят в колбу и концентрируют до объема 20-30 мл на водяной бане при температуре не выше 50° С в вакууме. К экстракту добавляют приблизительно равный объем 5%-ной спиртовой щелочи и омыляют в течение 30 мин-1 ч на водяной бане с обратным холодильником при кипении раствора. Омыленный раствор переносят в делительную воронку, прибавляют несколько миллилитров воды, взбалтывают и отделяют бензиновый слой, который затем промывают 8-10 раз дистиллированной водой. Бензиновый экстракт переносят в колбу и сушат обезвоженным сульфатом натрия при взбалтывании до исчезновения мутности раствора, затем фильтруют и концентрируют до объема 5-10 мл, как указано выше. Сгущенный экстракт пропускают при небольшом разрежении через адсорбционную колонку, наполненную окисью магния или окисью алюминия. Каротин, адсорбированный на колонке, элюируют (растворяют) эфиром или бензином, пропуская их через адсорбент до тех пор, пока выходящая из колонки жидкость не станет бесцветной.
Полученный фильтрат собирают в мерную колбу, доводят объем жидкости до метки петролейным эфиром или бензином и колориметрируют в колориметре Дюбоска или на фотоэлектроколориметре, используя для сравнения стандартный раствор азобензола или бихромата калия.
Второй вариант. Вначале проводят омыление исследуемого вещества, а затем экстрагирование каротина, адсорбцию и колориметрирование. Навеску измельченного вещества (от 1 до 50 г), растертую в ступке, переносят в колбу, прибавляют 20-40 мл 5%-ной спиртовой щелочи, омыляют в течение 30 мин-1 ч и дальше поступают так же, как и при первом способе.
Третий вариант (упрощенный). При этом способе исключается омыление, а все остальные стадии анализа те же, что и при первом способе.
Полученные экстракты промывают водой, сушат над безводным сернокислым натрием, концентрируют до малых объемов, пропускают через колонку с адсорбентом и колориметрируют.
При определении каротина в моркови можно исключить применение адсорбционной колонки, так как в моркови содержится незначительное количество других каротиноидов, которые практически мало влияют на результат определения. Анализ по третьему варианту проводят в тех случаях, когда результаты определения каротина совпадают с результатами, полученными при работе по первому варианту. Определение каротина в сухом растительном материале (овощи, плоды, ягоды и другие продукты). Навеску измельченного вещества берут от 2 до 10 г, каротин извлекают бензином или петролейным эфиром без предварительной обработки спиртом. Полученные экстракты сгущают до объема 20-30 мл и омыляют спиртовым раствором КОН. Далее анализ проводят, как указано в первом варианте.
Вычисление содержания каротина. При использовании для колориметрирования колориметра Дюбоска и стандартных растворов азобензола или бихромата калия содержание каротина (х) в мг % в исследуемом продукте рассчитывают по формуле
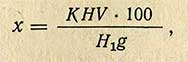
где К - коэффициент пересчета (количество каротина в миллиграммах, соответствующее 1 мл стандартного раствора азобензола, - 0,00235 или стандартного раствора биххромата калия 0,00208); H - показание шкалы стандартного раствора, мм; H1- показание шкалы испытуемого раствора, мм; g - навеска исследуемого продукта, г; V - объем фильтрата после хроматографической адсорбции, мл.
При использовании электрофотоколориметра применяют следующую формулу:
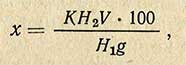
где H2 - показание шкалы реохорда для стандартного раствора; H1 - то же, для испытуемого раствора. Остальные обозначения такие же, как и в предыдущей формуле.
Приготовление стандартных растворов
Раствор азобензола. 14,5 мг кристаллического химически чистого азобензола растворяют в 100 мл 96%-ного этилового спирта.
Раствор бихромата калия. 360 мг трижды перекристаллизованного бихромата калия растворяют в 1 л дистиллированной воды.
Приготовление адсорбционной колонки
Для адсорбционной колонки используют стеклянную трубку длиной 12-15 см, диаметром 1-1,5 см, суженную книзу. Трубку вставляют через пробку в колбу Бунзена. В нижнюю часть адсорбционной трубки помещают вату, а затем адсорбент - окись магния или окись алюминия. Для этого приготовляют кашицу из адсорбента и бензина или петролейного эфира. Кашицей заполняют колонку на 4-6 см и промывают небольшими порциями растворителя, избегая образования пузырьков воздуха.
Определение витамина B1
Витамин B1 (тиамин, аневрин) находится в естественных продуктах как в свободном, так и в связанном виде. В первом случае - это свободный тиамин или его хлорид - гидрохлорид (C12H18O4Cl2); в связанном состоянии он представляет собой пирофосфорнокислый эфир тиамина, соединенный с белковым носителем, т.е. является коферментом карбоксилазы. В основу метода определения витамина B1, положена способность тиамина окисляться в тиохром феррицианидом калия в щелочной среде и свойство образовавшегося тиохрома давать голубую флуоресценцию при освещении ультрафиолетовыми лучами. В ходе анализа тиохром извлекают из водно-щелочного раствора изобутиловым, бутиловым или изоамиловым спиртом, отделяя его таким образом от флуоресцирующих и других нежелательных примесей, нерастворимых в указанных спиртах.
Содержание тиамина в исследуемом веществе устанавливают проводя на флуорометре сравнительное определение интенсивности флуоресценции испытуемого и стандартного растворов. Описанный метод применим для определения не только свободного тиамина, но и общего содержания тиамина. В этом случае связанную форму тиамина предварительно подвергают расщеплению ферментным препаратом, содержащим фосфатазу.
Флуорометрический метод определения витамина B1. Навеску исследуемого продукта в количестве 5-10 г, помещенную в ступку, тщательно растирают с 10-25 мл 0,1 н. раствора серной кислоты и переносят количественно в колбу при помощи того же раствора кислоты; общий объем жидкости в колбе доводят приблизительно до 75 мл. Колбу закрывают пробкой с обратным холодильником (воздушным), опускают в кипящую водяную баню и в течение 45 мин при периодическом перемешивании содержимого ее ведут экстракцию тиамина. В случае определения свободного тиамина полученную вытяжку охлаждают, добавляют 2,5 молярного раствора уксуснокислого натрия до pH 5,0, доводят объем до 100 мл дистиллированной водой, перемешивают, фильтруют и 10-20 мл раствора берут для дальнейшего анализа.
При определении общего содержания тиамина вытяжку охлаждают до 35-40° С и добавляют в нее ферментный препарат, который в количестве 0,03 г на 1 г сухого вещества навески предварительно растирают в ступке с 2-3 мл 2,5 молярного раствора уксуснокислого натрия, затем полученную взвесь препарата переносят в колбу при помощи 2-3 мл раствора уксуснокислого натрия и этим же раствором доводят pH вытяжки до 5,0.
Колбу с вытяжкой после добавления ферментного препарата закрывают ватной пробкой и помещают на 12-15 ч в термостат при температуре 37° С. Затем содержимое колбы охлаждают, доводят объем дистиллированной водой до 100 мл, перемешивают и фильтруют. Дальнейшее определение свободного тиамина и общего его содержания проводят одинаково.
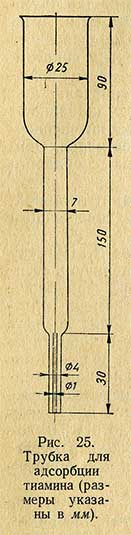
10-20 мл фильтрата пропускают через адсорбционную колонку для адсорбции тиамина. Для этой цели служит стеклянная трубка (рис. 25), имеющая следующие размеры: в верхней части - диаметр 25 мм и длину 90 мм, в средней части - диаметр 7 мм и длину 150 мм и в нижней части - диаметр 5 мм (внутренний диаметр 0,03-1,0 мм) и длину 30 мм. В среднюю часть трубки кладут стеклянную вату и сверху насыпают адсорбент; для катионита ОДВ-3 высота столбика должна быть около 8 см. Подготовленную к работе колонку укрепляют на пробке в мерном цилиндре емкостью 100 мл. Адсорбент промывают 10 мл 3%-ного раствора уксусной кислоты и пропускают через колонку испытуемый раствор. Затем адсорбент 3 раза промывают дистиллированной водой по 10 мл и элюируют тиамин с адсорбента нагретым до кипения 25%-ным раствором хлористого калия в 0,1 н. растворе соляной кислоты порциями по 6-7 мл. Элюат собирают в чистый градуированный цилиндр до объема 30 мл.
По 5 мл полученного раствора переносят пипеткой в две маленькие делительные воронки; в первую воронку добавляют 3 мл смеси для окисления тиамина (0,4%-ный раствор феррицианида калия в 15%-ном растворе едкого натра), перемешивают и приливают для извлечения образовавшегося тиохрома 12 мл изобутилового (бутилового или изоамилового) спирта. Во вторую воронку (контрольная проба) приливают 3 мл 15%-ного раствора едкого натра, перемешивают и добавляют 12 мл изобутилового спирта. Обе воронки встряхивают в течение 2 мин, оставляют смесь в покое до полного расслоения, отделяют нижний водно-щелочной слой, а спиртовой слой фильтруют через бумажный фильтр, в который предварительно помещают 2-3 г безводного сернокислого натрия; прозрачный фильтрат собирают в сухую пробирку, откуда его переносят в кювету флуорометра. Спиртовой раствор можно также обезвоживать сернокислым натрием непосредственно в делительной воронке; после внесения около 2 г реактива смесь встряхивают и обезвоженный раствор фильтруют через бумажный фильтр в сухую пробирку.
Раствор тиохрома из стандартного раствора тиамина готовят следующим образом: в две делительные воронки вносят градуированной пипеткой по 1 мл раствора, содержащего 1 мкг тиамина, добавляют по 4 мл 25%-ного раствора хлористого калия и затем в одну воронку приливают 3 мл смеси для окисления, а во вторую (контрольная проба) - 3 мл 15%-ного раствора едкого натра. Содержимое воронок перемешивают и добавляют в каждую воронку по 12 мл изобутилового спирта. В дальнейшем поступают, как описано выше.

Интенсивность флуоресценции подготовленных спиртовых растворов определяют на флуорометре (рис. 26) со специальными светофильтрами при помощи чувствительного гальванометра. Измеряют интенсивность флуоресценции в четырех растворах: в двух испытуемых (окисленном и контрольном неокисленном) и в двух стандартных (окисленном и контрольном неокисленном). В каждую кювету вносят около 8 мл изобутилового раствора.
Содержание тиамина в мг% (х) вычисляют по формуле
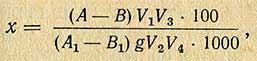
где A - показание флуорометра для испытуемого окисленного раствора; B - показание флуорометра для испытуемого неокисленного раствора; A1 - показание флуорометра для стандартного окисленного раствора; B1 - показание флуорометра для стандартного неокисленного раствора; g - навеска исследуемого продукта, г; V1 - общий объем вытяжки, мл; V2 - объем вытяжки, взятый для адсорбции, мл; V3 - общий объем элюата, мл; V4 - объем элюата, взятый для окисления, мл; 1000 - коэффициент пересчета, мг.
Приготовление основных реактивов и препаратов
1. Стандартный раствор тиамина. 10 мг кристаллического тиамин-хлорида растворяют в 0,001 н. 25%-ном спиртовом растворе соляной кислоты в мерной колбе емкостью 100 мл. Раствор не изменяется в течение 1-1,5 месяцев при хранении его в темной склянке в прохладном месте. Для приготовления рабочего раствора 1 мл стандартного раствора вносят в колбу емкостью 100 мл и разводят дистиллированной водой до метки; раствор готовят перед анализом, он содержит 1 мкг тиамина в 1 мл.
2. 2,5 молярный раствор уксуснокислого натрия. 340 г уксуснокислого натрия растворяют в дистиллированной воде и доводят объем до 1 л.
3. 25%-ный раствор хлористого калия. 250 г хлористого калия растворяют в дистиллированной воде, добавляют 8,5 мл концентрированной соляной кислоты и доводят объем водой до 1 л.
4. Смесь для окисления - 0,04%-ный раствор феррицианида калия в 15%-ном растворе едкого натра. Смесь готовят перед анализом, смешивая 4 мл свежеприготовленного 1 %-ного раствора феррицианида калия с 96 мл 15%-ного раствора едкого натрия.
5. Ферментные препараты из пенициллиума нотатум или из аспергиллуса ориза.
6. Адсорбент катионит СДВ-3. Катионит измельчают до размера частиц от 0,5 до 0,13 мм в количестве 70% и менее 0,13 мм - 30%. Для освобождения от примесей железа обрабатывают его троекратно 10%-ной соляной кислотой каждый раз по 2 ч при 40-60° С, промывают дистиллированной водой до исчезновения реакции на хлор и активируют подсушиванием при температуре не выше 60-70° С.
Определение витамина B2
Витамин B2 (рибофлавин) C17H20N4O6 содержится в естественных продуктах как в свободном, так и в связанном состоянии. Известны три формы связанного рибофлавина: флавинмононуклеотид, флавинадениндинуклеотид и третья форма, прочно связанная с белком.
Метод определения витамина B2 основан на свойстве водных растворов рибофлавина давать интенсивную желто-зеленую флуоресценцию в ультрафиолетовом свете. При определении общего содержания витамина В2 флуорометрическим методом рибофлавин связанных форм переводят в свободное состояние ферментативным и кислотным гидролизом. В ходе анализа вытяжки из естественных продуктов обрабатывают последовательно перманганатом и гидросульфитом натрия для уменьшения количества флуоресцирующих примесей. Затем в отдельной пробе определяют интенсивность неспецифической флуоресценции, которая зависит только от оставшихся примесей; в этой пробе предварительно восстанавливают рибофлавин в бесцветную лейкоформу и таким образом «гасят» его флуоресценцию. При расчете содержания витамина B2 в исследуемом продукте данные по неспецифической флуоресценции вводят как поправку в результат определения общей флуоресценции.
Определения общего содержания витамина B2. Навеску продукта (5-10 г) тщательно растирают в ступке с небольшим количеством фосфатного буфера (pH 7,8-8,0), после чего переносят в колбу при помощи того же буферного раствора, доводя общее разведение до соотношения 1:15 или 1:20. Колбу с содержимым нагревают на кипящей водной бане в течение 45 мин при частом перемешивании, охлаждают до 30° С, проверяют величину pH и в случае сдвига в кислую зону снова доводят pH до 7,8-8,0 добавлением фосфатного буфера. К вытяжке добавляют ферментный препарат (трипсин, панкреатин или препарат из пенициллиума нотатум) в количестве 30 мг на 1 г сухого вещества навески, который предварительно растирают в ступке с 2-3 мл фосфатного буфера или уксуснокислого натрия. Вытяжку выдерживают в термостате при 37° С в течение 12-20 ч; при ферментативном гидролизе отщепляется прочно связанная с белком форма рибофлавина. После охлаждения вытяжку доводят дистиллированной водой до объема, соответствующего общему разведению 1:25 или 1:30, и фильтруют через складчатый фильтр.
В небольшую колбу вносят 5 мл фильтрата, приливают 5 мл 20%-ного трихлоруксусной кислоты и нагревают на кипящей водяной бане в течение 10 мин. Раствор охлаждают и добавляют 1/4 объема 4-молярного раствора двузамещенного фосфата калия для доведения величины pH до 6,0. Затем к вытяжке приливают по каплям 4%-ный раствор перманганата для окисления флуоресцирующих примесей; раствор перманганата прибавляют обычно в количестве 0,2-0,4 мл до появления стойкой красноватой окраски вытяжки.
Вытяжку, обработанную перманганатом, оставляют в покое на 10 мин, а затем к ней приливают по каплям 3%-ный раствор перекиси водорода до тех пор, пока не исчезнет окраска; при добавлении перекиси водорода вытяжку непрерывно взбалтывают. К вытяжке прибавляют 0,2 мл рабочего раствора хлористого олова и 0,1 мл 2,5%-ного раствора гидросульфита натрия для восстановления флуоресцирующих примесей. Вытяжку энергично встряхивают в течение 20 мин для перевода обратимо восстановленного рибофлавина в окисленную флуоресцирующую форму. Объем вытяжки доводят водой до 15 мл, при наличии мути раствор фильтруют. В подготовленной вытяжке определяют интенсивность флуоресценции по сравнению с интенсивностью флуоресценции стандартного рабочего раствора рибофлавина. Для этого вытяжку и рабочий раствор рибофлавина (см. ниже «приготовление реактивов») наливают по 8-10 мл в кюветы флуорометра и измеряют интенсивность флуоресценции по шкале гальванометра. Далее в обе кюветы добавляют по 0,1 г кислого углекислого натрия и по 0,1 г гидросульфита, перемешивают содержимое кювет и снова измеряют интенсивность флуоресценции. В стандартном растворе рибофлавина флуоресценция гасится до нуля, а в исследуемой вытяжке сохраняется небольшая флуоресценция, что обусловлено наличием флуоресцирующих примесей, которые не удаляются полностью при обработке вытяжки указанными выше реагентами. Чтобы убедиться в полноте гашения флуоресценции рибофлавина, к пробам добавляют по 0,1 г гидросульфита и снова измеряют интенсивность флуоресценции. При полном гашении показания гальванометра не должны изменяться. Содержание рибофлавина в микрограммах на 1 г веществ (х) вычисляют по формуле
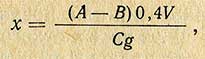
где А - показание флуорометра для испытуемого раствора (первый отсчет); В - показание флуорометра для испытуемого раствора после гашения (второй отсчет); С - показание флуорометра для стандартного раствора, содержащего 0,4 мкг рибофлавина в 1 мл; 0,4 - концентрация стандартного раствора, мкг; g - навеска продукта, г; V - объем общего разведения, мл.
Приготовление основных реактивов
1. Стандартный раствор рибофлавина. Навеску рибофлавина в количестве 10 мг растворяют в дистиллированной воде в мерной колбе емкостью 250 мл. 1 мл такого раствора содержит 40 мкг рибофлавина. Раствор не изменяется в течение 1 месяца при хранении на холоду и в темноте. Перед определением приготовляют рабочий раствор, для чего в мерную колбу емкостью 100 мл вносят 37,5 мл 20%-ного раствора трихлоруксусной кислоты, 25 мл 4-молярного раствора двузамещенного фосфата калия, 1 мл стандартного раствора рибофлавина и доводят водой до метки. 1 мл рабочего раствора содержит 0,4 мкг рибофлавина.
2. Фосфатная буферная смесь (pH 7,8-8,0). Приготовляют 1/15-молярный раствор двузамещенного фосфата натрия (11,876 г перекристаллизованного Na2HPO4-2H2O в 1 л воды) и 1/15-молярный раствор однозамещенного фосфата калия (9,078 г перекристаллизованного КН2РO4 в 1 л воды). Смешивают 9,5 части первого раствора и 0,5 части второго раствора.
3. Раствор хлористого олова. 10г хлористого олова (SnCl2) растворяют в 25 мл концентрированной соляной кислоты. Полученный основной раствор хранят в темной склянке с притертой пробкой при комнатной температуре. Перед каждым определением готовят рабочий раствор разбавлением 0,2 мл основного раствора водой до 100 мл.
4. Раствор гидросульфита натрия. 0,25 г Na2S2O4-2Н2O растворяют в 10 мл 2%-ного раствора двууглекислого натра. Раствор готовят перед употреблением.
5. Ферментные препараты: трипсин, панкреатин или ферментный препарат из пенициллиума нотатум.
Определение никотиновой кислоты (витамина PP)
В естественных продуктах витамин РР (никотиновая кислота) встречается в свободном и связанном виде: как никотиновая кислота C6H5O2N или ее амид C6H6ON2. Для определения никотиновой кислоты предложен колориметрический метод, который основан на взаимодействии никотиновой кислоты с бромистым роданидом или цианом. Образующееся при этом соединение в присутствии ароматических аминов (анилин, метол) в нейтральной или слабокислой среде дает производное, окрашенное в желтый цвет. Интенсивность окраски испытуемых растворов прямо пропорциональна количеству никотиновой кислоты и измеряется колориметрически.
Методика определения. Навеску измельченного исследуемого продукта берут в количестве 5 г, переносят в мерную колбу емкостью 100 мл и приливают 75 мл 2-н. раствора серной кислоты, смывая воронку и горлышко колбы раствором этой кислоты. Содержимое колбы энергично перемешивают. Колбу помещают в кипящую водяную баню и нагревают содержимое в течение 90 мин при периодическом перемешивании. После этого колбу охлаждают, доводят смесь до метки дистиллированной водой, тщательно перемешивают и фильтруют через бумажный фильтр. (Полученный гидролизат можно оставить на холоду до следующего дня).
Берут 25 мл фильтрата, помещают в мерную колбу емкостью 50 мл, добавляют одну каплю фенолфталеина и вносят 10 н. раствор едкого натра до получения слабо-розового окрашивания (примерно 4 мл). Избыток щелочи устраняют 1-2 каплями 5 н. серной кислоты (до исчезновения розового окрашивания). Если раствор нагрелся, его охлаждают, а затем добавляют 2 мл раствора сернокислого цинка и 1-2 капли изоамилового спирта (для устранения пены). Затем при перемешивании содержимого колбы добавляют по каплям раствор 4 н. едкого натра до образования густого осадка гидроокиси цинка. Осаждение заканчивают добавлением раствора 1 н. едкого натра до появления бледно-розового окрашивания. В колбу добавляют 1-2 капли 5 н. серной кислоты (до исчезновения розового окрашивания) и оставляют стоять в течение 10 мин при периодическом помешивании. Смесь в колбе доводят до 50 мл дистиллированной водой, перемешивают и фильтруют через бумажный фильтр. Полученный фильтрат используют для проведения цветных реакций, для этого применяют специальные пробирки с пришлифованными пробками, которые вставляют в штатив круглой формы. Одновременно при проведении цветных реакций испытуемых растворов аналогичные операции повторяют со стандартными растворами никотиновой кислоты. При этом ставят контроль на реактивы к стандартным растворам и на амины к испытуемым.
Перечень растворов, используемых при проведении анализа, приведен в табл. 5.
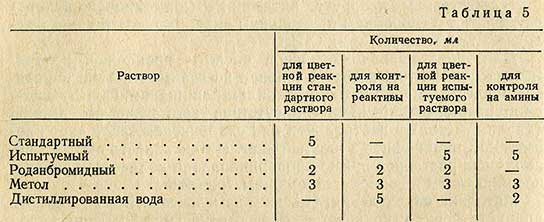
Для проведения цветных реакций в две пробирки (параллельные определения) приливают по 5 мл стандартного раствора никотиновой кислоты и в две пробирки по 5 мл дистиллированной воды, затем в четыре другие пробирки приливают по 5 мл испытуемого раствора. Все пробирки, помещенные в штатив, погружают в баню при температуре 50° С на 5 мин, после чего под тягой из бюретки добавляют по 2 мл роданбромидного раствора согласно табл. 5 (исключая контроль на амины). Жидкость в пробирках перемешивают и оставляют их в бане на 10 мин при температуре 50° С. Пробирки охлаждают в холодной воде до комнатной температуры, помещают в деревянный ящичек с гнездами для пробирок, закрывают ящик крышкой и оставляют стоять в темном месте в течение 10 мин. В пробирки добавляют по 3 мл раствора метола, содержимое перемешивают и оставляют в закрытом ящике на 1 ч в темном месте.
По истечении часа полученные растворы колориметрируют на фотоэлектроколориметре при синем светофильтре в кювете при толщине слоя 10 мм. Содержание никотиновой кислоты вычисляют следующим образом. Устанавливают величины оптической плотности испытуемого (n) и стандартного (n1) растворов с учетом поправок на контроль
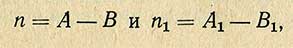
где А - оптическая плотность испытуемого раствора; А1 - то же, стандартного; В - оптическая плотность контрольного раствора на амины; B1 - оптическая плотность контрольного раствора на реактивы.
В дальнейшем для расчета содержания никотиновой кислоты в мг% (x) используют следующую формулу:
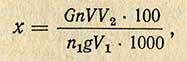
где G - содержание никотиновой кислоты в 1 мл стандартного раствора, мгк; n - оптическая плотность испытуемого раствора с учетом контрольного раствора; n1 - оптическая плотность стандартного раствора с учетом контрольного раствора; g - навеска, г; V - общий объем гидролизата, мл; V1 - объем гидролизата, взятый для очистки сернокислым цинком, мл; V2 - конечный объем раствора после добавления сернокислого цинка, мл.
Приготовление реактивов
1. Стандартный раствор никотиновой кислоты (основной). 500 мг никотиновой кислоты помещают в колбу емкостью 500 мл, добавляют 5 мл 10 н. H2SO4 и, когда кристаллы растворятся, доводят до метки дистиллированной водой. 1 мл такого раствора содержит 1000 мкг никотиновой кислоты. Раствор пригоден в течение 1 года при хранении на холоде.
2. Стандартный раствор - рабочий. 5 мл основного стандартного раствора разбавляют до 1 л дистиллированной водой. 1 мл такого раствора содержит 5 мкг никотиновой кислоты (раствор приготовляют ежедневно).
3. Роданбромидный раствор (готовят перед употреблением). Приготовляют бромную воду, внося в дистиллированную воду бром до прекращения растворения капель брома. К охлажденной на льду бромной воде, взятой в количестве необходимом для анализа, прибавляют по каплям 10%-ный раствор роданистого калия или аммония до светло-желтого окрашивания, а затем 1 %-ный раствор тех же реактивов до полного обесцвечивания бромной воды. Добавляют постепенно, небольшими порциями, по 20-50 мг углекислого кальция до прекращения выделения пузырьков и образования мути. Раствор фильтруют в склянку из темного стекла с притертой пробкой и хранят на холоде.
4. Раствор метола 8%-ный (приготовляют перед употреблением). 8 г перекристаллизованного метола растворяют в 0,5 н. растворе НСl и переносят в мерный цилиндр или колбу емкостью 100 мл, раствор доводят до метки 0,5 н. НСl.
Перекристаллизация метола. 500 мл 0,1 н. H2SO4 нагревают до кипения, в кипящий раствор добавляют 100 г метола, предварительно смешанного с 0,7 г NaHSO3; смесь нагревают до кипения. Если раствор сильно окрашен, добавляют 10 г активированного угля. Смесь немедленно переносят на предварительно нагретую воронку Бюхнера и фильтруют. В химический стакан переносят фильтрат, добавляют 0,3 г бисульфита натрия и 700 мл 96%-ного спирта; все перемешивают, погружают в ледяную воду и оставляют в темном месте на несколько часов. Выпавшие кристаллы метола фильтруют через Бюхнеровскую воронку, промывают на воронке 96%-ным спиртом из пульверизатора и высушивают на воздухе в темноте. Перекристаллизованный метол хранят в склянке из темного стекла с притертой пробкой в темном месте.